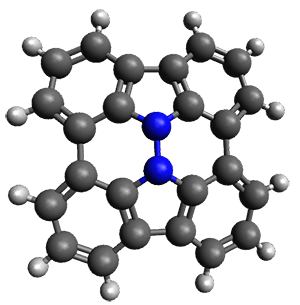
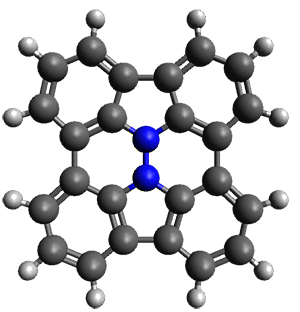
Cyclopropyl rings can be joined together in a spiro fashion to form triangulanes. An interesting topology can be made by joining the rings to form a helical pattern, as shown in the [9]triangulane 1 below. Allen, Quanz, and Schreiner1 have examined the notion of an infinite helical molecule formed in this way.
|
|
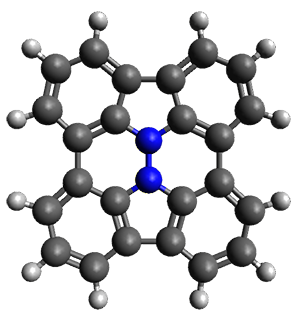
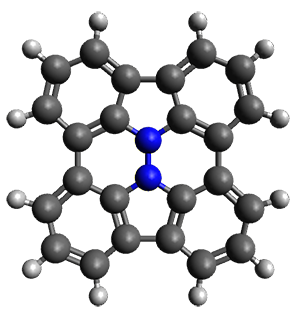
First, they describe how one can generate the coordinates of such a beast using a closed analytical expression, which is a really nice demonstration of applied geometry. Next, they compute the geometry of a series of [n]triangulanes at M06-2x/6-31G(d). The geometries of [9]triangulane and their largest example, [42]triangulane 2 are shown in Figure 1.
1 |
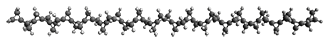
2 |
Figure 1. M06-2x/6-31G(d) optimized geometries of 1 and 2.
They show that the geometry of 2 exhibits a structure that has two different C-C distances: one between the spiro carbons, and the second between the spiro carbon and the methylene carbon. The distance between the spiro carbons is rather short (1.458 Å), suggesting that the bonding here is between carbons that are nearly sp2-hybridized.
Lastly, they discuss the thermodynamics of polytriangulane. They employ a series of homodesmotic reactions to attempt to determine the enthalpy for adding another cyclopropyl ring to an extended triangulane. Unfortunately, the computed enthalpy is quite dependent on functional used. Similar attempts to define the strain energy is also flawed in this way. However, regardless of the functional the enthalpy for adding a cyclopropane ring appears to reach an asymptote rather quickly. So, using [3]triangulane they estimate that the strain energy per mole of cyclopropane in triangulane is about 42.7 kcal mol-1, or about 14 kcal mol-1 of strain due to the spiroannulation.
References
(1) Allen, W. D.; Quanz, H.; Schreiner, P. R. “Polytriangulane,” J. Chem. Theory Comput. 2016, 12, 4707–4716, DOI: 10.1021/acs.jctc.6b00669.
InChIs
1: InChI=1S/C19H22/c1-2-12(1)5-14(12)7-16(14)9-18(16)11-19(18)10-17(19)8-15(17)6-13(15)3-4-13/h1-11H2/t14-,15-,16-,17-,18-,19-/m0/s1
InChIKey=XBTZCZDSKVTALB-DYKIIFRCSA-N