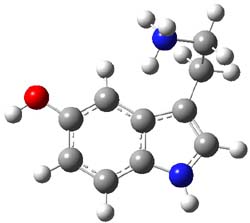
Palau’amine has been of interest since its discovery in the early 1990s. It was just recently synthesized by Baran,1 to much acclaim. The structure of palau’amine underwent numerous revisions, and though the relative configuration had been settled, the absolute configuration was only determined by Reinscheid and Griesinger using a combination of experimental and computed ECD and ORD spectra.2
|
|
3,4-dibromopalau’amine 1 was subjected to careful NMR analysis to set as much of the overall structure as possible. Then two conformations were optimized at B3LYP/6-31G(d), one of which is displayed in Figure 1.
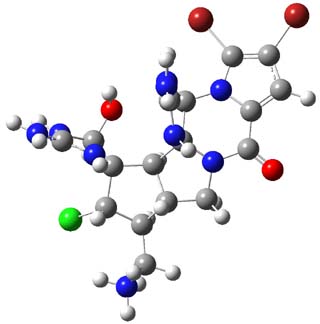 1 |
Figure 1. B3LYP/6-31G(d) optimized structure of 1.
TD-DFT computations including PCM gave an ECD spectrum that nicely matches with experiment, especially where the positive and negative peaks occur. The computed and experimental ORD spectra also match well, with all the signs matching up and a difference in the absolute value of the rotation of no more that 25%. The resulting absolute configuration is (-)-(6S,10R,11S,12S,16R,17S,18S,20S)-dibromopalau’amine, demonstrating again the power of combining computation and experiment for structure determination!
References
(1) Seiple, I. B.; Su, S.; Young, I. S.; Lewis, C. A.; Yamaguchi, J.; Baran, P. S., "Total Synthesis of Palau’amine," Angew. Chem. Int. Ed., 2010, 49, 1095-1098, DOI: 10.1002/anie.200907112
(2) Reinscheid, U. M.; Köck, M.; Cychon, C.; Schmidts, V.; Thiele, C. M.; Griesinger, C., "The Absolute Configuration of Dibromopalau’amine," Eur. J. Org. Chem., 2010, 6900-6903, DOI: 10.1002/ejoc.201001392
InChI
1: InChI=1/C17H22Br2ClN9O2/c18-6-1-7-11(30)28-3-5-4(2-21)9(20)16(13(31)25-15(23)26-16)8(5)17(28)12(24-14(22)27-17)29(7)10(6)19/h1,4-5,8-9,12-13,24-27,31H,2-3,21-23H2/q+2/p+1/t4-,5-,8+,9+,12+,13+,16+,17-/m1/s1/fC17H23Br2ClN9O2/h21H/q+3
InChIKey=VGQTUXLYZXIYTJ-CAGSSHLPDN