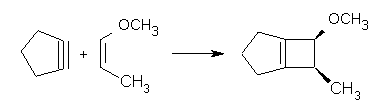
A nice follow-up to some of my own work points out again the possible dramatic role of dynamic effects. Way back when, Jack Gilbert discovered that the reaction of cyclopentyne with alkenes gives the cyclobutene product with stereoretention (Reaction 1),1 seemingly in violation of the Woodward-Hoffmann rules.
|
|
Reaction 1
|
Jack and I proposed an intermediate spirocyclopropyl carbene which could then open to product, and this would follow a stereoretention path.2,3 In a subsequent paper,4 we noted that a diradical pathway is also possible, and conjectured that dynamics might account for the stereoretention – that formation of the diradical leads directly to the carbene, leaving a very short lifetime of the diradical (Scheme 1). The consequence of the short lived diradical is that there little opportunity to rotate about the C-C bond and scramble the stereochemistry.
Scheme 1
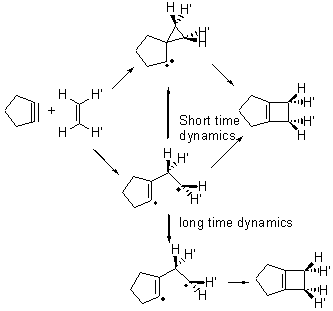
Pilling has published a MD study of this system and finds what we predicted.5 The short-time trajectories lead to stereoretention product. This is due to both passages over the TS that lead from the diradical to the product (with no scrambling) and over the TS that connects the diradical to the carbine. Longer trajectories do exhibit some stereoscrambling. Carpenter6 has argued that short time dynamics are often what one observes for potential energy surfaces like this one. Pilling also argues that in solution, with the actual alkene which bears bulky substituents that the proton (he examined the reaction of cyclopentyne with ethene), rotations will be slower, leading to formation of the carbene with stereoretention.
References
(1) Gilbert, J. C.; Baze, M. E., "Stereochemistry of [2 + 2] cycloadditions of cyclopentyne," J. Am. Chem. Soc. 2002, 106, 1885-1886, DOI: 10.1021/ja00318a081
(2) Laird, D. W.; Gilbert, J. C., "Norbornyne: A Cycloalkyne Reacting Like A Dicarbene," J. Am. Chem. Soc., 2001, 123, 6704-6705, DOI: 10.1021/ja010589h
(3) Bachrach, S. M.; Gilbert, J. C.; Laird, D. W., "DFT Study of the Cycloaddition Reactions of Strained Alkynes," J. Am. Chem. Soc., 2001, 123, 6706-6707, DOI: 10.1021/ja010590g
(4) Bachrach, S. M.; Gilbert, J. C., "The Reaction of Cyclopentyne with Ethene: Concerted vs Stepwise Mechanism?," J. Org. Chem., 2004, 69, 6357-6364, DOI: 10.1021/jo0492970
(5) Glowacki, D. R.; Marsden, S. P.; Pilling, M. J., "Significance of Nonstatistical Dynamics in Organic Reaction Mechanisms: Time-Dependent Stereoselectivity in Cyclopentyne−Alkene Cycloadditions," J. Am. Chem. Soc. 2009, 131, 13896-13897, DOI: 10.1021/ja9043054
(6) Barry, K. C., "Nonexponential decay of reactive intermediates: new challenges for spectroscopic observation, kinetic modeling and mechanistic interpretation," J. Phys. Org. Chem., 2003, 16, 858-868, DOI: 10.1002/poc.672