In sort of an afterword to a recent publication, Stanger1 points out an error made by Hückel in arguing for stability of D6h benzene over the hypothetical D3h cyclohexatriene. Hückel constructed the first two matrices shown below to describe each molecule. The energy of benzene, predicted by this matrix, is 6α-8β, which is lower than that for cyclohexatriene, 6α-6β.
Stanger points out that implicit in Hückel’s argument is that the value of the Hij element for the double bond is identical in value for the two compounds, (namely β), and the element is zero for the single bonds. Considering that the C-C double bond in cyclohexatriene should be shorter than that in benzene, Stanger suggests that its Hij element should be larger than β and the Hij element for the single bond should not be zero, but some small value reflecting the overlap between the p-orbitals. He suggests that the values for the single and double bond elements should be 0.4589β and 1.5050β, giving the bottom matrix below. This leads to an electronic energy of 6α-9.212β. In other words, this more appropriate model of cyclohexatriene has a lower π energy than does D6h benzene! This is in accord with Shaik’s argument2 that the π system of benzene acts to localize the bonds and it’s the σ system that is responsible for its delocalized structure.
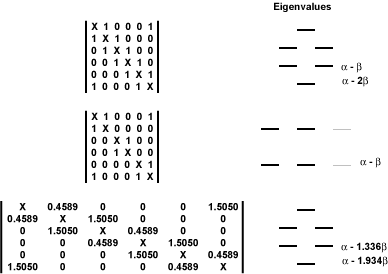
Figure 1. Hückel matrices and eigenvalues for benzene (top),
traditional cyclohexatriene model (middle), and revised cyclohexatriene model (bottom).1
References
(1) Stanger, A., "The Different Aromatic Characters of Some Localized Benzene Derivatives†," J. Phys. Chem. A, 2008, 112, 12849-12854, DOI: 10.1021/jp801634x
(2) Shaik, S.; Shurki, A.; Danovich, D.; Hiberty, P. C., "A different story of benzene," Journal of Molecular Structure: THEOCHEM, 1997, 398-399, 155-167, DOI: 10.1016/S0166-1280(96)04934-2.

Henry Rzepa responded on 17 Feb 2009 at 12:10 pm #
Perhaps I am missing something here, but surely one has to compare these results with an appropriate reference state. For benzene, this would be three separate molecules of ethene. If the resonance integral for the “short” bond in cyclohexatriene is around 1.5β, then surely for the even shorter bond in ethene it might be around 2.0β. This would give ethene a total energy of 2α + 4β, or for three of them, 6α + 12β. This makes the benzene almost 3 β units less stable than three ethenes. Even if one assigned the resonance integral in ethene to the same as the shorter bond in the modified treatment, it would give a total energy of 6α + 9β for three ethenes. Again, benzene by this criterion has no real stabilization compared to three ethenes. I presume then that it could only be the σ framework that can be providing the resonance stabilization for benzene?