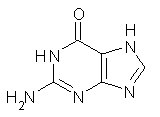
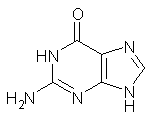
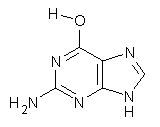
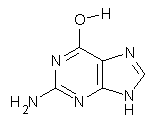
As mentioned in Chapter 2 of my book, many post-HF methods predict that planar benzene has an imaginary frequency, whereby out-of-plane bending leads to a lower energy structure.1 This anomaly was suggested to result from intramolecular basis set incompleteness.
Asturiol, Duran and Salvador provide more evidence that the root cause is intramolecular basis set superposition error.2 They propose an extension of the standard counterpoise correction, which has been widely applied to interacting molecules. They divide the molecule into small fragments and apply the counterpoise correction to these fragments. For benzene, they use C-H or (CH)2 fragments. With this counterpoise correction, the imaginary frequency corresponding to an out-of-plane distortion is removed for all combinations of either MP2 or CISD with the 6-31+G*, 6-311G or 6-311++G basis sets. The planar indenyl anion, which is found to have 4 imaginary frequencies at MP2/6-311G, has no imaginary frequencies when the counterpoise correction is used.
These authors have now shown that nucleic acid bases suffer from the same intramolecular superposition error.3 Uracil, thymine and guanine suffer from spurious imaginary frequencies with certain combinations of MP2 and Pople basis sets. However, all of these out-of-plane imaginary frequencies become real when the counterpoise correction is applied. The take-home message is to carefully mate the post-HF method and basis set combination – or else make the counterpoise correction!
References
(1) Moran, D.; Simmonett, A. C.; Leach, F. E.; Allen, W. D.; Schleyer, P. v. R.; Schaefer, H. F., III, "Popular Theoretical Methods Predict Benzene and Arenes To Be Nonplanar," J. Am. Chem. Soc. 2006, 128, 9342-9343, DOI: 10.1021/ja0630285
(2) Asturiol, D.; Duran, M.; Salvador, P., "Intramolecular basis set superposition error effects on the planarity of benzene and other aromatic molecules: A solution to the problem," J. Chem. Phys. 2008, 128, 144108, DOI: 10.1063/1.2902974
(3) Asturiol, D.; Duran, M.; Salvador, P., "Intramolecular Basis Set Superposition Error Effects on the Planarity of DNA and RNA Nucleobases," J. Chem. Theory Comput. 2009, 5, 2574-2581, DOI: 10.1021/ct900056u