I know this post is off topic for the blog, but yesterday’s New York Times simply raised my blood pressure.
The Book Review of the New York Times on Sunday March 30 has a review on the book Bonk. The book discusses sex research – and while that certainly is of interest – I want to focus on the associated artwork.
Now chemistry is not usually considered a particularly sexy subject, but its graphics can be fascinating. The periodic table might be the most widely known scientific graphic. The structure of DNA has captured the imagination of more than just scientists. And so it’s not unreasonable that the Times Book Review editors would choose to present some chemical (2-D) drawings. Given the subject of the book, one might have expected perhaps the structures of testosterone and progesterone. Instead, we get the following:
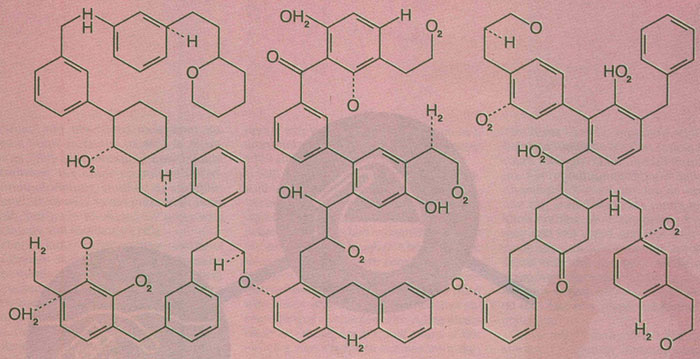
While these structures do not correspond to any known compound – not in and of itself a bad sin – the knowledge (or lack thereof) of chemistry displayed here is simply amazing. The graphic designer is certainly drawn to the preponderance of 6-member rings found in organic chemistry, but has taken this to an extreme! It seems that a random collection of atomic symbols were then willy-nilly added wherever the artist thought would be interesting. The result is simply absurd. One could only have hoped that some copy-editor with a chemistry background could have noticed some of the more glaring mistakes – oh, like five bonds to carbon! For the newspaper of record, these mistakes are simply unwarranted.
To make something good out of this, I am going to hold a contest with my first-semester organic students to identify the errors. The winner will get extra credit in the class – and I will post results here later on.