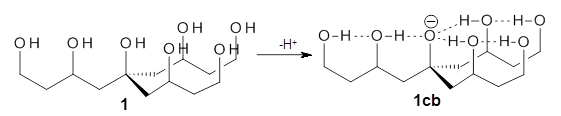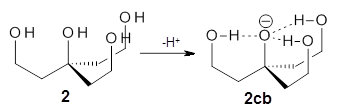Kass and coworkers looked at a series of substituted phenols to tease out ways to produce stronger acids in non-polar media.1 First they established a linear relationship between the vibrational frequency shifts of the hydroxyl group in going from CCl4 as solvent to CCl4 doped with 1% acetonitrile with the experimental pKa in DMSO. They also showed a strong relationship between this vibrational frequency shift and gas phase acidity (both experimental and computed deprotonation energies).
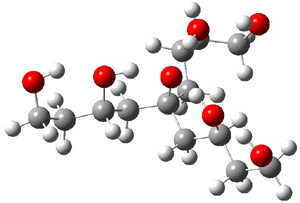
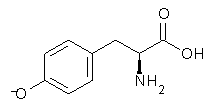
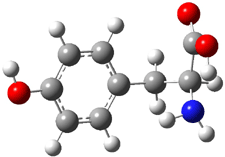
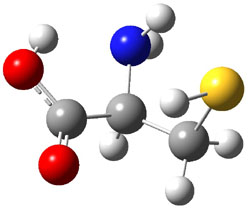
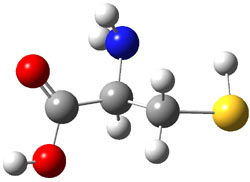
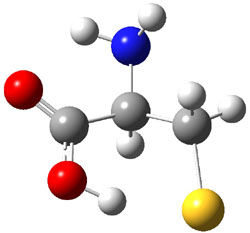
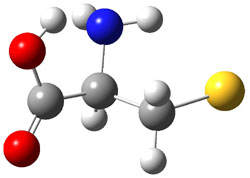
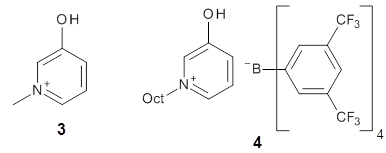
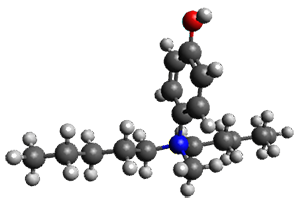
A key recognition was that a charged substituent (like say ammonium) has a much larger effect on the gas-phase (and non-polar solvent) acidity than on the acidity in a polar solvent, like DMSO. This can be attributed to the lack of a medium able to stable charge build-up in non-polar solvent or in the gas phase. This led them to 1, for which B3LYP/6-31+G(d,p) computations of the analogous dipentyl derivative 2 (see Figure 1) indicated a deprotonation free energy of 261.4 kcal mol-1, nearly 60 kcal mol-1 smaller than any other substituted phenol they previously examined. Subsequent measurement of the OH vibrational frequency shift showed the largest shift, indicating that 1 is extremely acidic in non-polar solvent.
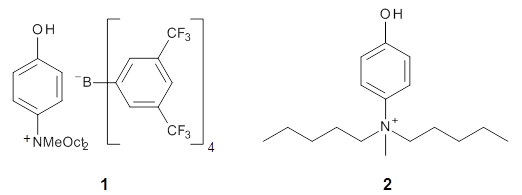
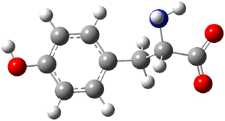
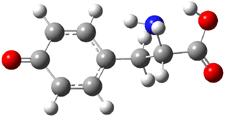
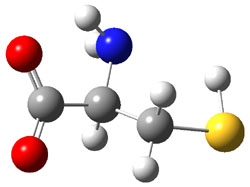
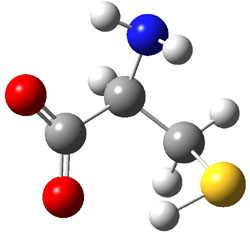
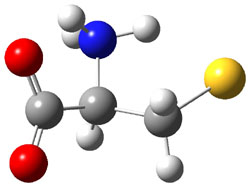
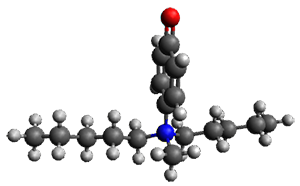
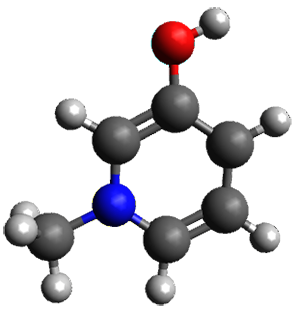
Further computational exploration led to 3 (see Figure 1), for which computations predicted an even smaller deprotonation energy of 231.1 kcal mol-1. Preparation of 4 and experimental observation of its vibrational frequency shift revealed an even larger shift than for 1, making 4 extraordinarily acidic.
2 |
Conjugate base of 2 |
 3 |
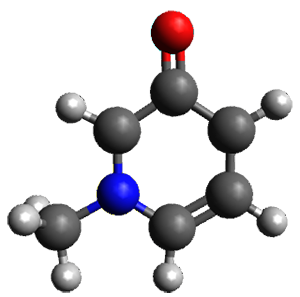 Conjugate base of 3 |
Figure 1. B3LYP/6-31+G(d,p) optimized geometries of 2 and 3 and their conjugate bases.
Reference
(1) Samet, M.; Buhle, J.; Zhou, Y.; Kass, S. R. "Charge-Enhanced Acidity and Catalyst Activation," J. Am. Chem. Soc. 2015, 137, 4678-4680, DOI: 10.1021/jacs.5b01805.
InChI
1 (cation only): InChI=1S/C23H41NO/c1-4-6-8-10-12-14-20-24(3,21-15-13-11-9-7-5-2)22-16-18-23(25)19-17-22/h16-19H,4-15,20-21H2,1-3H3/p+1
InChIKey=HIQMXPFMEWRQQG-UHFFFAOYSA-O
2:
3: InChI=1S/C6H7NO/c1-7-4-2-3-6(8)5-7/h2-5H,1H3/p+1
InChIKey=FZVAZYLFYPULKX-UHFFFAOYSA-O
4 (cation only): InChI=1S/C13H21NO/c1-2-3-4-5-6-7-10-14-11-8-9-13(15)12-14/h8-9,11-12H,2-7,10H2,1H3/p+1
InChIKey=HSFRKOBOATYXAH-UHFFFAOYSA-O