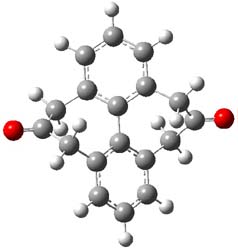
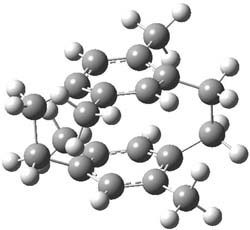
I have written a number of blog posts that deal with the computation of optical activity. Trindle and Altun have now reported TD-DFT computations of circular dichroism of high-symmetry molecules.1 The employ either B3LYP (with a variety of basis sets, the largest being 6-311++G(2d,2p)) and SOAP/ATZP. For a number of the high symmetry molecules (two examples are shown in Figure 1), the two methods differ a bit in their predictions of the first excited state, with SOAP typically predicting a red shift relative to the B3LYP. However, both methods general give the same sign of the CD signals and their line shapes are similar.
|
|
|
 |
 |
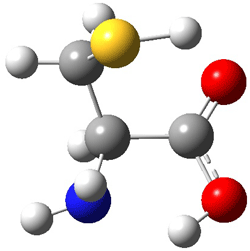 I |
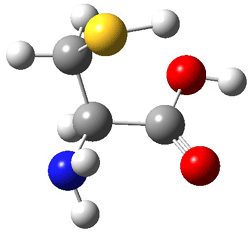 II |
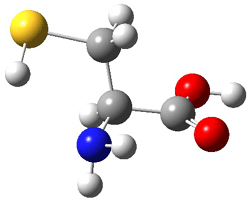 III |
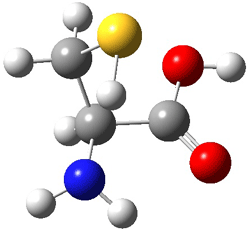 IV |
Figure 1. MP2(FC)/aug-cc-pV(T+d)Z optimized geometries and focal point relative energies (kJ mol-1) of the four lowest energy conformers of cysteine.1
The three lowest energy structures found here match up with the lowest two structures found by Alonso and the energy differences are also quite comparable: 4.79 kJ and 5.81 mol-1 with the focal point method 3.89 and 5.38 kJ mol-1 with MP4/6-311++G(d,p)// MP2/6-311++G(d,p). So the identification of the cysteine conformers made by Alonso remains on firm ground.
References
(1) Wilke, J. J.; Lind, M. C.; Schaefer, H. F.; Csaszar, A. G.; Allen, W. D., "Conformers of Gaseous Cysteine," J. Chem. Theory Comput. 2009, DOI: 10.1021/ct900005c.
(2) Sanz, M. E.; Blanco, S.; López, J. C.; Alonso, J. L., "Rotational Probes of Six Conformers of Neutral Cysteine," Angew. Chem. Int. Ed. 2008, 4, 6216-6220, DOI: 10.1002/anie.200801337
InChIs
Cysteine:
InChI=1/C3H7NO2S/c4-2(1-7)3(5)6/h2,7H,1,4H2,(H,5,6)/t2-/m0/s1
InChIKey: XUJNEKJLAYXESH-REOHCLBHBU
amino acids &focal point &Schaefer Steven Bachrach 13 Jul 2009 1 Comment
Hexaporphyrin that’s Möbius aromatic
The Kim and Osuka groups have reported another Möbius aromatic porphyrin, 1, a 28 π-electron system.1 This hexaporphyrin is produced without the need for low temperature, complexation with a metal or protonation (see this post for a discussion of their earlier work). The x-ray crystal structure shows the Möbius twist, and the 1H NMR shifts of the interior protons at 2.22 and 1.03 ppm. B3LYP/6-31G** computations indicate a NICS value at the center of the molecule of -11.8 ppm. These are consistent with aromatic behavior.
|
|
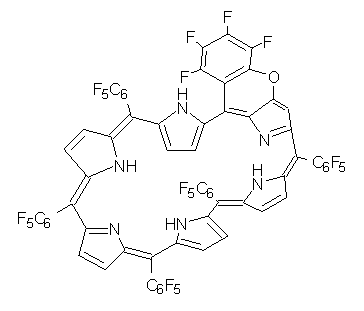
References
(1) Tokuji, S.; Shin, J.-Y.; Kim, K. S.; Lim, J. M.; Youfu, K.; Saito, S.; Kim, D.; Osuka, A., "Facile Formation of a Benzopyrane-Fused [28]Hexaphyrin That Exhibits Distinct Möbius Aromaticity," J. Am. Chem. Soc. 2009, 131, 7240-7241, DOI: 10.1021/ja902836x.
InChIs
1: InChIKey=YGJLOPZWRVMFIJ-XFAHNSIYBC
Aromaticity Steven Bachrach 07 Jul 2009 No Comments
-
Categories
- Acidity (12)
- Aromaticity (91)
- Authors (153)
- Bond Dissociation Energy (6)
- BSSE (1)
- cyclophane (0)
- Dynamics (35)
- E-publishing (7)
- Enzyme (4)
- FEP (1)
- host-guest (6)
- Hydrogen bond (5)
- Ion Pairs (1)
- Isotope Effects (5)
- Keto-enol tautomerization (3)
-
Molecules (100)
- adamantane (3)
- amino acids (13)
- annulenes (8)
- benzynes (4)
- biphenyl (1)
- calixarenes (1)
- carbenes (13)
- cyclobutadiene (4)
- dendralenes (1)
- Dewar benzene (1)
- diradicals (8)
- ephedrine (1)
- ethyl cation (2)
- fullerene (6)
- fulvalenes (1)
- hexacyclinol (2)
- nanohoops (4)
- non-classical (4)
- norbornyl cation (2)
- nucleic acids (4)
- oximes (1)
- phenyloxenium (1)
- polycyclic aromatics (7)
- propellane (2)
- stilbene (1)
- sugars (5)
- terpenes (2)
- twistane (1)
- NMR (40)
- Optical Rotation (16)
-
QM Method (96)
- CASPT2 (1)
- DFT (71)
- focal point (7)
- G3 (3)
- MP (11)
-
Reactions (83)
- 1,2-addition (1)
- aldol (4)
- Bergman cyclization (6)
- Claisen rearrangement (2)
- Cope Rearrangement (5)
- cycloadditions (12)
- Diels-Alder (26)
- electrocyclization (11)
- electrophilic aromatic substitution (1)
- ene reaction (1)
- Hajos-Parrish Reaction (1)
- Mannich (2)
- Michael addition (5)
- ozonolysis (1)
- proton transfer (1)
- pseudopericyclic (4)
- Strecker (1)
- Substitution (6)
- Wittig (1)
- Second Edition (3)
- Solvation (17)
- Stereochemistry (2)
- stereoinduction (4)
- Tunneling (26)
- Uncategorized (57)
- vibrational frequencies (3)
-
Monthly
- June 2019
- April 2019
- March 2019
- February 2019
- January 2019
- December 2018
- November 2018
- October 2018
- September 2018
- August 2018
- July 2018
- June 2018
- May 2018
- April 2018
- March 2018
- February 2018
- January 2018
- December 2017
- November 2017
- October 2017
- September 2017
- August 2017
- July 2017
- June 2017
- May 2017
- April 2017
- March 2017
- February 2017
- January 2017
- December 2016
- November 2016
- October 2016
- September 2016
- August 2016
- July 2016
- June 2016
- May 2016
- April 2016
- March 2016
- February 2016
- January 2016
- December 2015
- November 2015
- October 2015
- September 2015
- August 2015
- July 2015
- June 2015
- May 2015
- April 2015
- March 2015
- February 2015
- January 2015
- December 2014
- November 2014
- October 2014
- September 2014
- August 2014
- July 2014
- June 2014
- May 2014
- April 2014
- March 2014
- February 2014
- January 2014
- December 2013
- November 2013
- October 2013
- September 2013
- August 2013
- July 2013
- June 2013
- May 2013
- April 2013
- March 2013
- February 2013
- January 2013
- December 2012
- November 2012
- October 2012
- September 2012
- August 2012
- July 2012
- June 2012
- May 2012
- April 2012
- March 2012
- February 2012
- January 2012
- December 2011
- November 2011
- October 2011
- September 2011
- August 2011
- July 2011
- June 2011
- May 2011
- April 2011
- March 2011
- February 2011
- January 2011
- December 2010
- November 2010
- October 2010
- September 2010
- August 2010
- July 2010
- June 2010
- May 2010
- April 2010
- March 2010
- February 2010
- January 2010
- December 2009
- November 2009
- October 2009
- September 2009
- August 2009
- July 2009
- June 2009
- May 2009
- April 2009
- March 2009
- February 2009
- January 2009
- December 2008
- November 2008
- October 2008
- September 2008
- August 2008
- July 2008
- June 2008
- May 2008
- April 2008
- March 2008
- February 2008
- January 2008
- December 2007
- November 2007
- October 2007
- September 2007
- August 2007
- July 2007

This work is licensed under a Creative Commons Attribution-No Derivative Works 3.0 Unported License.