Often gem-dialkyl substitution accelerates a reaction, for example in the formation of an epoxide via reaction 1. Here the relative rates are 1:21:252 in going from 1 to 2 to 3.1 This acceleration is the Thorpe-Ingold effect and had been suggested to arise from a steric reaction: that the methyl groups contract the angle and bring the terminal groups closer together.
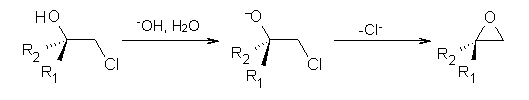
1: R1 = R2 = H
2: R1 = Me, R2 = H
3: R1 = R2 = Me
Kostal and Jorgensen2 have examined the reaction of the 2-chloroethoxides 1-3 using computations, especially to look at the effect of solvent. At MP2/6-311+G(d,p) and CBS-Q, the relative rates (based on the activation free energy ΔG‡) are 1:2.8:17 and 1:0.7:3.7, respectively. Evidently there is no significant rate enhancement afforded by gem-substitution in the gas phase.
However, solution computations give a very different result. Using PCM along with the MP2 method, the computed relative rates are 1:5.8:1100 and with the Monte Carlo-Free Energy Perturbation method, the relative rates for aqueous solution are 1:30:773. Thus, the Thorpe-Ingold acceleration is due to solvent. Analysis of the hydrogen bonded structures and the solute-water pair distributions suggest that increasing alkyl substitution reduces the strength of solvation of the reactant, leading to the lower activation barrier.
References
(1) Jung, M. E.; Piizzi, G., "gem-Disubstituent Effect:Theoretical Basis and Synthetic Applications," Chem. Rev., 2005, 105, 1735-1766, DOI: 10.1021/cr940337h
(2) Kostal, J.; Jorgensen, W. L., "Thorpe-Ingold Acceleration of Oxirane Formation Is Mostly a Solvent Effect," J. Am. Chem. Soc., 2010, 132, 8766-8773, DOI: 10.1021/ja1023755