Hobza1 has published a very high-level computational study of the benzene dimer as a benchmark for this model of π-π stacking – a topic I have touched upon a number of times in this blog (post 1, post 2) . There are four local energy minima, shown in Figure 1. The most stable dimer is the tilted T-structure (TT), a structure often overlooked. Its complexation energy, computed at CCSD(T)/CBS, is 2.78 kcal mol-1. Only slightly higher in energy is the parallel displaced structure (PD), with a stabilization energy of 2.70 kcal mol-1. The T structure (T) is essentially isoenergetic with the PD one. The perfectly stacked structure (S) is much less stable, with a dimerization energy of 1.64 kcal mol-1. The DTF-D method, using the BLYP functional with dispersion parameters optimized for the benzene dimer provide energies within 0.2 kcal mol-1 of the computationally much more expensive benchmark values. As a word of caution though: use of more general dispersion parameters gives energies far worse and predicts the wrong energy order of the dimers.
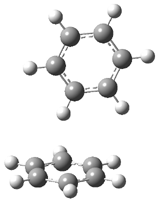 TT |
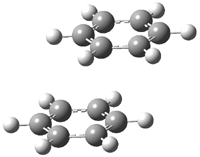 PD |
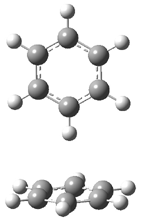 T |
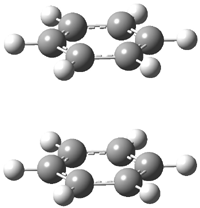 S |
Figure 1. Structures of the benzene dimer with stabilization energy (kcal mol-1) computed at CCSD(T)/CBS (bold), DFT-D/BLYP with optimized parameters (italics), and DFT-D/BLYP with general parameters (plain).1
References
(1) Pitonak, M.; Neogrady, P.; Rexac, J.; Jurecka, P.; Urban, M.; Hobza, P., "Benzene Dimer: High-Level Wave Function and Density Functional Theory Calculations," J. Chem. Theory Comput., 2008, 4, 1829-1834, DOI: 10.1021/ct800229h.

Paulo E. Abreu responded on 29 Jan 2009 at 12:19 pm #
Hiya
Have you seen this article on benzene dimer “Geometries and stabilities of various configurations of benzene dimer: details of novel V-shaped structure revealed” http://www.springerlink.com/content/r7408575134w1g36/ (DOI: 10.1007/s11224-009-9411-6)
I was just reading my feeds when I found this article. Although I didnt have the time to read it thoroughly it seems to have new and interestings results.
Thanks for your blog 🙂
Paulo
sbachrach responded on 29 Jan 2009 at 12:36 pm #
Thanks Paulo for this link. I have not read it yet, but I will since it is relevent to the blog theme.
Steven
Ashutosh responded on 05 Feb 2009 at 1:26 pm #
Hi Steven
Great blog and great book. I was wondering if you are aware of an experimental and theoretical value for the hydration energy of the benzene dimer. I want to know this number for a proposal I am writing and cannot seem to find it anywhere. if you are aware of a reference, I would really appreciate it if you can email me at ajogale@emory.edu.
Thanks very much!
Ashutosh