Schreiner and Grimme have examined a few compounds (see these previous posts) with long C-C bonds that are found in congested systems where dispersion greatly aids in stabilizing the stretched bond. Their new paper1 continues this theme by examining 1 (again) and 2, using computations, and x-ray crystallography and gas-phase rotational spectroscopy and electron diffraction to establish the long C-C bond.
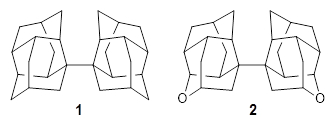
The distance of the long central bond in 1 is 1.647 Å (x-ray) and 1.630 Å (electron diffraction). Similarly, this distance in 2 is 1.642 Å (x-ray) and 1.632 Å (ED). These experiments discount any role for crystal packing forces in leading to the long bond.
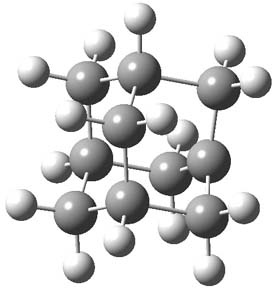
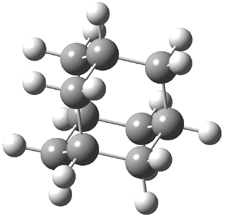
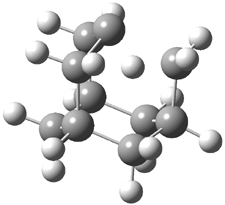
A very nice result from the computations is that most functionals that include some dispersion correction predict the C-C distance in the optimized structures with an error of no more than 0.01 Å. (PW6B95-D3/DEF2-QZVP structures are shown in Figure 1.) Not surprisingly, HF and B3LYP without a dispersion correction predict a bond that is too long.) MP2 predicts a distance that is too short, but SCS-MP2 does a very good job.
|
|
|
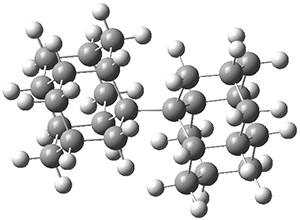
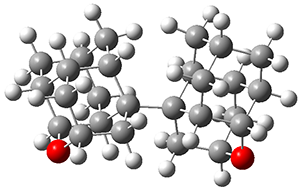
Figure 1. PW6B95-D3/DEF2-QZVP optimized structures of 1 and 2.
References
1) Fokin, A. A.; Zhuk, T. S.; Blomeyer, S.; Pérez, C.; Chernish, L. V.; Pashenko, A. E.; Antony, J.; Vishnevskiy, Y. V.; Berger, R. J. F.; Grimme, S.; Logemann, C.; Schnell, M.; Mitzel, N. W.; Schreiner, P. R., "Intramolecular London Dispersion Interaction Effects on Gas-Phase and Solid-State Structures of Diamondoid Dimers." J. Am. Chem. Soc. 2017, 139, 16696-16707, DOI: 10.1021/jacs.7b07884.
InChIs
1: InChI=1S/C28H38/c1-13-7-23-19-3-15-4-20(17(1)19)24(8-13)27(23,11-15)28-12-16-5-21-18-2-14(9-25(21)28)10-26(28)22(18)6-16/h13-26H,1-12H2
InChIKey=MMYAZLNWLGPULP-UHFFFAOYSA-N
2: InChI=1S/C26H34O2/c1-11-3-19-15-7-13-9-25(19,21(5-11)23(27-13)17(1)15)26-10-14-8-16-18-2-12(4-20(16)26)6-22(26)24(18)28-14/h11-24H,1-10H2
InChIKey=VPBJYHMTINJMAE-UHFFFAOYSA-N