Can vibrational spectroscopy be used to identify stereoisomers? Medel, Stelbrink, and Suhm have examined the vibrational spectra of (+)- and (-)-α-pinene, (±)-1, in the presence of four different chiral terpenes 2-5.1 They recorded gas phase spectra by thermal expansion of a chiral α-pinene with each chiral terpene.
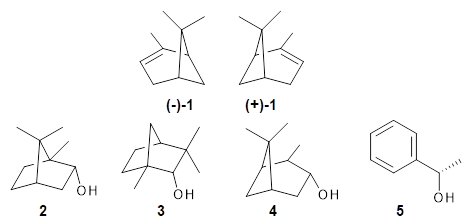
For the complex of 4 with (+)-1 or (-)-1 and 5 with (+)-1 or (-)-1, the OH vibrational frequency is identical for the two different stereoisomers. However, the OH vibrational frequencies differ by 2 cm-1 with 3, and the complex of 3/(+)-1 displays two different OH stretches that differ by 11 cm-1. And in the case of the complex of α-pinene with 2, the OH vibrational frequencies of the two different stereoisomers differ by 11 cm-1!
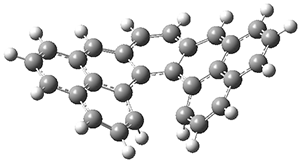
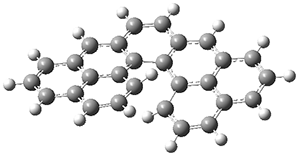
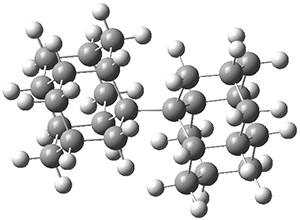
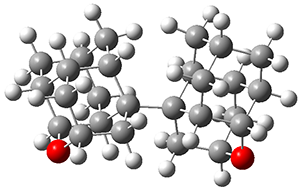
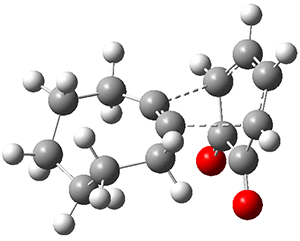
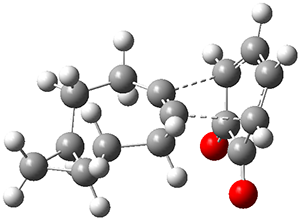
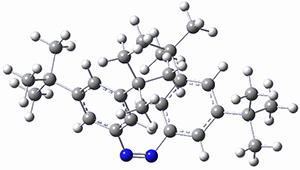
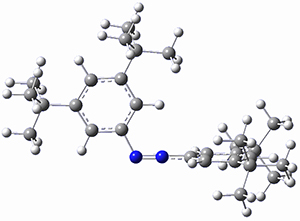
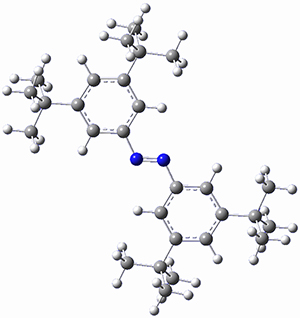
The B3LYP-D3(BJ)/def2-TZVP optimized geometry of the 2/(+)-1 and 2/(-)-1 complexes are shown in Figure 2, and some subtle differences in sterics and dispersion give rise to the different vibrational frequencies.
|
|
|
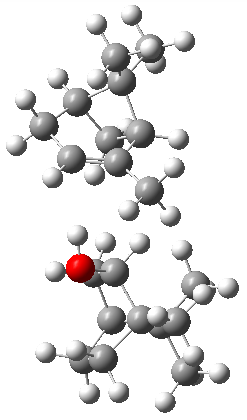
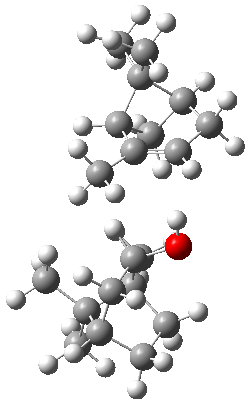
Figure 2. B3LYP-D3(BJ)/def2-TZVP optimized geometry of the 2/(+)-1 and 2/(-)-1
Of interest to readers of this blog will be the DFT study of these complexes. The authors used three different well-known methods – B3LYP-D3(BJ)/def2-TZVP, M06-2x/def2-TZVP, and ωB97X-D/def2-TZVP – to compute structures and (most importantly) predict the vibrational frequencies. Interestingly, M06-2x/def2-TZVP and ωB97X-D/ def2-TZVP both failed to predict the vibrational frequency difference between the complexes with the two stereoisomers of α-pinene. However, B3LYP-D3(BJ)/def2-TZVP performed extremely well, with a mean average error (MAE) of only 1.9 cm-1 for the four different terpenes. Using this functional and the larger may-cc-pvtz basis set reduced the MAE to 1.5 cm-1 with the largest error of only 2.5 cm-1.
As the authors note, these complexes provide some fertile ground for further experimental and computational study and benchmarking.
Reference
1. Medel, R.; Stelbrink, C.; Suhm, M. A., “Vibrational Signatures of Chirality Recognition Between α-Pinene and Alcohols for Theory Benchmarking.” Angew. Chem. Int. Ed. 2019, 58, 8177-8181, DOI: 10.1002/anie.201901687.
InChIs
(-)-1, (-)-α-pinene: InChI=1S/C10H16/c1-7-4-5-8-6-9(7)10(8,2)3/h4,8-9H,5-6H2,1-3H3/t8-,9-/m0/s1
InChIKey=GRWFGVWFFZKLTI-IUCAKERBSA-N
(+)-1, (-)-α-pinene: InChI=1S/C10H16/c1-7-4-5-8-6-9(7)10(8,2)3/h4,8-9H,5-6H2,1-3H3/t8-,9-/m1/s1
InChIKey=GRWFGVWFFZKLTI-RKDXNWHRSA-N
2, (-)borneol: InChI=1S/C10H18O/c1-9(2)7-4-5-10(9,3)8(11)6-7/h7-8,11H,4-6H2,1-3H3/t7-,8+,10+/m0/s1
InChiKey=DTGKSKDOIYIVQL-QXFUBDJGSA-N
3, (+)-fenchol: InChI=1S/C10H18O/c1-9(2)7-4-5-10(3,6-7)8(9)11/h7-8,11H,4-6H2,1-3H3/t7-,8-,10+/m0/s1
InChIKey=IAIHUHQCLTYTSF-OYNCUSHFSA-N
4, (-1)-isopinocampheol: InChI=1S/C10H18O/c1-6-8-4-7(5-9(6)11)10(8,2)3/h6-9,11H,4-5H2,1-3H3/t6-,7+,8-,9-/m1/s1
InChIKey=REPVLJRCJUVQFA-BZNPZCIMSA-N
5, (1S)-1-phenylethanol: InChI=1S/C8H10O/c1-7(9)8-5-3-2-4-6-8/h2-7,9H,1H3/t7-/m0/s1
InChIKey=WAPNOHKVXSQRPX-ZETCQYMHSA-N