We have all learned about aromatic substitution as proceeding via the following mechanism
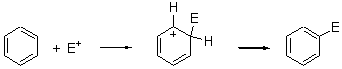
(Worse yet – many of us have taught this for years!) Well, Galabov, Zou, Schaefer and Schleyer pour a whole lot of cold water on this notion in their recent Angewandte article.1 Modeling the reaction of benzene with Br2 and using B3LYP/6-311+G(2d,2p) for both the gas phase and PCM simulating a CCl4 solvent, attempts to locate this standard intermediate led instead to a concerted substitution transition state TS1 (see Figure 1).
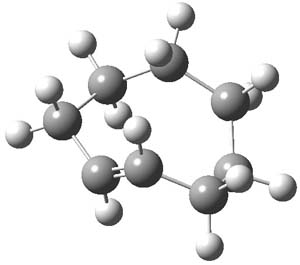
 TS1 |
Figure 1. PCM/B3LYP/6-311+G(2d,2p) optimized transitin state along the concerted pathway
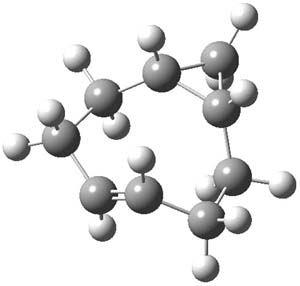
However, this is not the lowest energy pathway for substitution. Rather and addition-elimination pathway is kinetically preferred. In the first step Br2 adds in either a 1,2 or 1,4 fashion to form an intermediate. The lower energy path is the 1,4 addition, leading to P3. This intermediate then undergoes a syn,anti-isomerization to give P5. The last step is the elimination of HBr from P5 to give the product, bromobenzene. This mechanism is shown in Scheme 2 and the critical points are shown in Figure 3.
Scheme 1

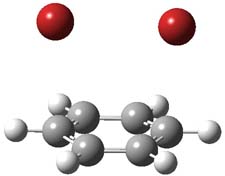 TS3 |
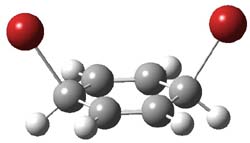 P3 |
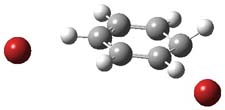 TS6 |
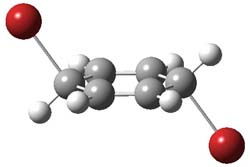 P5 |
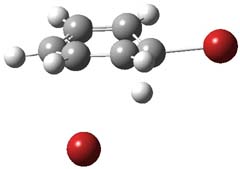 TS9 |
|
Figure 2. PCM/B3LYP/6-311+G(2d,2p) optimized critical points along the addition-elimination pathway
The barrier for the concerted substitution process through TS1 is 41.8 kcal mol-1 (in CCl4) while the highest barrier for the addition-elimination process is through TS3 of 39.4 kcal mol-1.
Now a bit of saving grace is that in polar solvents, acidic solvents and/or with Lewis acid catalysts, the intermediate of the standard textbook mechanism may be competitive.
Textbook authors – please be aware!
References
(1) Kong, J.; Galabov, B.; Koleva, G.; Zou, J.-J.; Schaefer, H. F.; Schleyer, P. v. R., "The Inherent Competition between Addition and Substitution Reactions of Br2 with Benzene and Arenes," Angew. Chem. Int. Ed. 2011, 50, 6809-6813, DOI: 10.1002/anie.201101852