In solution ortho-phenylenes preferentially coil into a helix with the phenyl rings stacked. However, 25-50% of these chains will typically misfold. Hartley and coworkers have reported the use of substituents to increase the percentage of perfectly folded chains.1
They synthesized two isomeric o-phenylenes, differing in the substitution pattern (1 and 2), with chain length of 6 to 10 phenyl rings. Substituents included methoxy, acetoxy, nitrile, and triflate. They principally employed 1H NMR to assess the conformational distribution, and used computations to confirm the conformation.
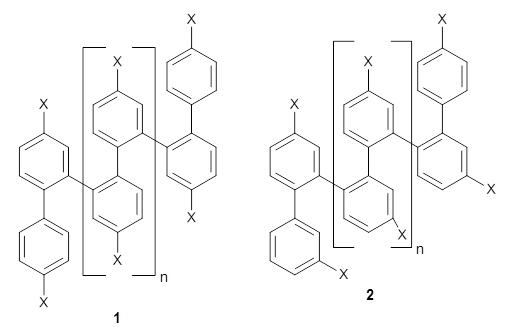
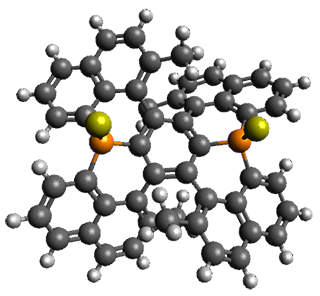
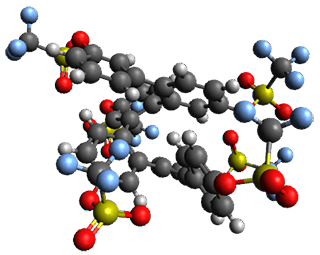
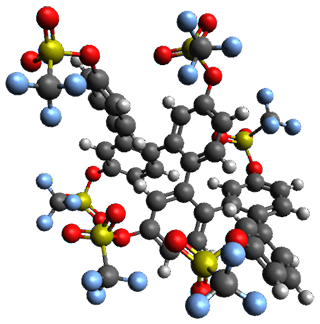
Ideally folded conformations of 1 and 2 with eight phenyl rings are shown in Figure 1. The dihedral angle formed by two adjacent phenyl rings are typically about ±55° or ±130°.
Figure 1. Idealized folding of 1 and 2 with X=OH.
Hydrogens omitted in these images, but full structures available, through Jmol, by clicking on the image.)
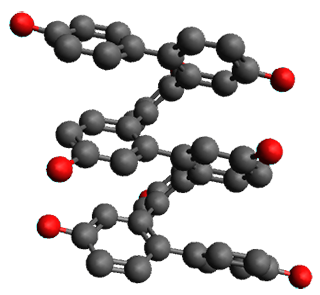
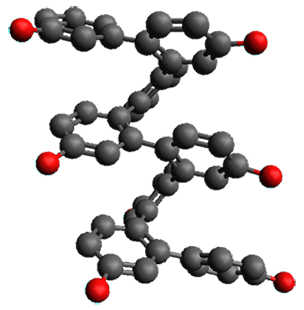
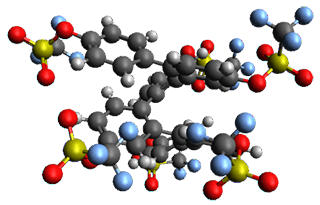
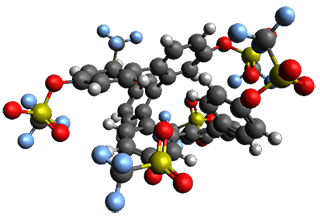
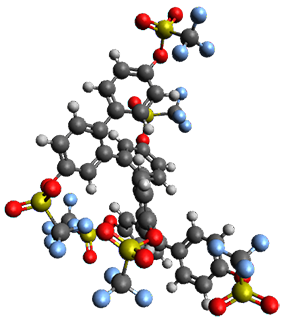
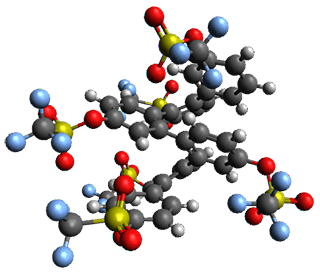
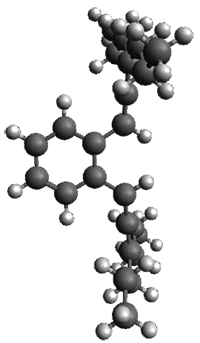
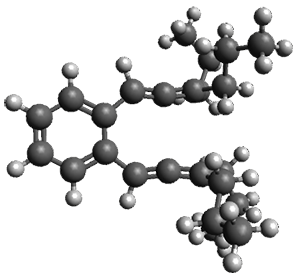
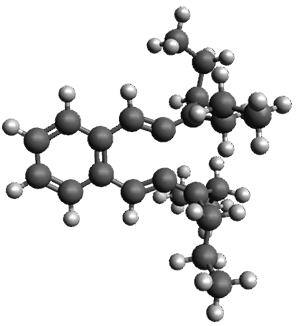
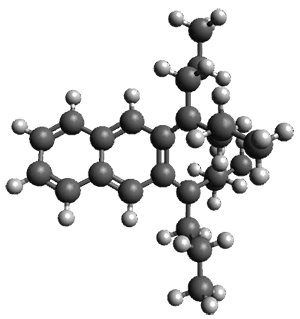
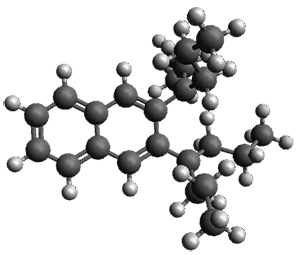
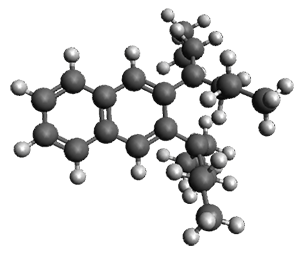
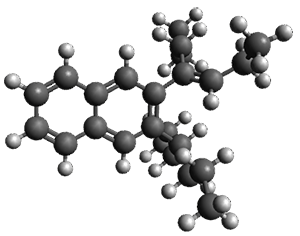
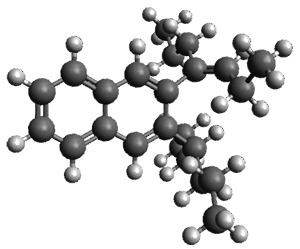
Given the size of these systems, and the conformation flexibility not just of the chain but with each substituent, a full search to identify the global minimum was not undertaken. Rather, a library of conformations was generated with MM, the lowest 200 conformations were then reoptimized at PM7 and then the energies were determined at PCM/B97-D/TZV(2d,2p). The lowest energy conformer was then reoptimized at this DFT level. Three conformations of 3 and 4 are shown in Figure 2 with triflate as the substituent with six phenyl rings. The first conformer has optimal stacking (perfect folding), the second conformer as one misfold at the end, and the third conformer has no stacking at all.
|
3
– ideal fold
|
3
– one misfold
|
|
3 – all misfold
|
|
4 – ideal fold
|
4
– one misfold
|
|
4
– all misfold
|
Figure 2. Optimized geometries of conformers of 3 and 4.
(Remember that clicking on one of these images will bring up the JMol applet allowing you to rotate and visualize the molecule in 3-D – a very useful feature here!)
NMR chemical shifts were then computed using these geometries at PCM/WP04/6-31G(d). In all cases examined, the chemical shifts of the major conformation was confirmed to be the perfect folding one by comparison with the computed chemical shifts. The examined substituents enhanced the proportion of properly folded chains in all cases, often to the extent where no minor conformer was observed at all.
References
(1) Mathew, S.; Crandall, L. A.; Ziegler, C. J.; Hartley, C. S. "Enhanced Helical Folding of ortho-Phenylenes through the Control of Aromatic Stacking Interactions," J. Am. Chem. Soc. 2014, 136, 16666-16675, DOI:10.1021/ja509902m.
InChIs
3: InChI=1S/C42H26F18O18S6/c43-37(44,45)79(61,62,63)23-5-1-21(2-6-23)29-13-9-25(81(67,68,69)39(49,50)51)17-33(29)35-19-27(83(73,74,75)41(55,56)57)11-15-31(35)32-16-12-28(84(76,77,78)42(58,59)60)20-36(32)34-18-26(82(70,71,72)40(52,53)54)10-14-30(34)22-3-7-24(8-4-22)80(64,65,66)38(46,47)48/h1-20H,(H,61,62,63)(H,64,65,66)(H,67,68,69)(H,70,71,72)(H,73,74,75)(H,76,77,78)
InChIKey=BOCNJIZJHQKUNK-UHFFFAOYSA-N
4: InChI=1S/C42H26F18O18S6/c43-37(44,45)79(61,62,63)23-6-4-21(5-7-23)33-17-25(81(67,68,69)39(49,50)51)9-13-30(33)35-19-27(83(73,74,75)41(55,56)57)11-15-32(35)36-20-28(84(76,77,78)42(58,59)60)10-14-31(36)34-18-26(82(70,71,72)40(52,53)54)8-12-29(34)22-2-1-3-24(16-22)80(64,65,66)38(46,47)48/h1-20H,(H,61,62,63)(H,64,65,66)(H,67,68,69)(H,70,71,72)(H,73,74,75)(H,76,77,78)
InChIKey=YKSIXAVDGFBUIZ-UHFFFAOYSA-N