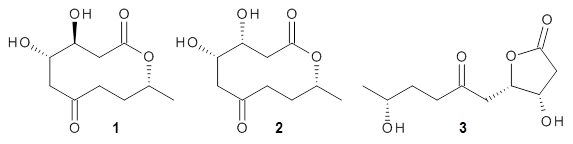
I have posted on the use of computed NMR chemical shifts and coupling constants to help aid in structure identification. The second edition of my book Computational Organic Chemistry has a largely all-new chapter on structure identification aided by computed spectra, especially NMR spectra. In my recent opinion piece speculating on challenges in computational organic chemistry,1 the first area I highlight is encouraging the larger use of computed spectra as an essential component of structure determination.
While more and more non-traditional computational users are employing quantum computations towards these problems, I suspect that many non-users are a bit wary about stepping into an arena they are not expert in, an arena chock-filled with acronyms and methods and potentially little guidance. While some very nice papers2-6 and web sites (Chemical Shift Repository (Cheshire) and DP4) do outline procedures for using computations in this fashion, they are not truly designed for the non-specialist.
Well, fear not any longer. Hoye and coworkers, synthetic chemists who have utilized computational approaches in structure determinations for a number of years, have written a detailed step-by-step protocol for using a standard computational approach towards structure determination.7 The article is written with the synthetic chemist in mind, and includes a number of scripts to automate many of the steps.
For the specialist, the overall outline of the protocol is fairly routine:
- Utilize MacroModel to perform a conformational search for each proposed structure, retaining the geometries within 5 kcal mol-1 of the global minimum.
- Optimize these conformations for each structure at M06-2x/6-31+G(d).
- For each conformation of each structure, compute the 1H and 13C chemical shifts, scale them, and determine the Boltzmann weighted chemical shifts
- Compare these chemical shifts with the experimental values using Mean Absolute Error
The article is straightforward and easily guides the novice user through these steps. Anyone unsure of how to utilize quantum chemical computations in structure determination is well advised to start with this article.
References
(1) Bachrach, S. M. "Challenges in computational organic chemistry," WIRES: Comput. Mol. Sci. 2014, 4, 482-487, DOI: 10.1002/wcms.1185.
(2) Lodewyk, M. W.; Siebert, M. R.; Tantillo, D. J. "Computational Prediction of 1H and 13C Chemical Shifts: A Useful Tool for Natural Product, Mechanistic, and Synthetic Organic Chemistry," Chem. Rev. 2012, 112, 1839–1862, DOI: 10.1021/cr200106v.
(3) Bally, T.; Rablen, P. R. "Quantum-Chemical Simulation of 1H NMR Spectra. 2. Comparison of DFT-Based Procedures for Computing Proton-Proton Coupling Constants in Organic Molecules," J. Org. Chem. 2011, 76, 4818-4830, DOI: 10.1021/jo200513q.>
(4) Jain, R.; Bally, T.; Rablen, P. R. "Calculating Accurate Proton Chemical Shifts of Organic Molecules with Density Functional Methods and Modest Basis Sets," J. Org. Chem. 2009, 74, 4017-4023, DOI: 10.1021/jo900482q.
(5) Smith, S. G.; Goodman, J. M. "Assigning the Stereochemistry of Pairs of Diastereoisomers Using GIAO NMR Shift Calculation," J. Org. Chem. 2009, 74, 4597-4607, DOI: 10.1021/jo900408d.
(6) Smith, S. G.; Goodman, J. M. "Assigning Stereochemistry to Single Diastereoisomers by GIAO NMR Calculation: The DP4 Probability," J. Am. Chem. Soc. 2010, 132, 12946-12959, DOI: 10.1021/ja105035r.
(7) Willoughby, P. H.; Jansma, M. J.; Hoye, T. R. "A guide to small-molecule structure assignment through computation of (1H and 13C) NMR chemical shifts," Nat. Protocols 2014, 9, 643-660, DOI: 10.1038/nprot.2014.042.