Concern about the use of DFT for general use in organic chemistry remains high; see my previous posts (1, 2, 3). Houk has now examined the reaction enthalpies of ten simple Diels-Alder reactions using a variety of functionals in the search for the root cause of the problem(s).1
The ten reactions are listed in Scheme 1, and involve cyclic and acyclic dienes and either ethylene or acetylene as the dienophile. Table 1 lists the minimum and maximum deviation of the DFT enthalpies relative to the CBS-QB3 enthalpies (which are in excellent accord with experiment). Clearly, all of the DFT methods perform poorly, with significant errors in these simple reaction energies. The exception is the MO6-2X functional, whose errors are only slightly larger than that found with the SCS-MP2 method. Use of a larger basis set (6-311+G(2df,2p)) reduced errors only a small amount.
|
Scheme 1 |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
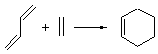
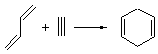
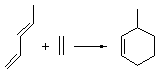
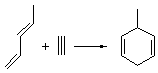
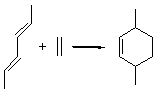
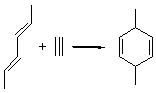
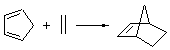
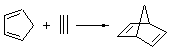
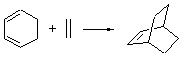
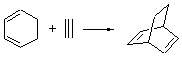
Table 1. Maximum, minimum and mean deviation of reaction enthalpies (kcal mol-1) for the reactions in Scheme 1 using the 6-31+G(d,p) basis set.1
|
Method |
Maximum Deviation |
Minimum Deviation |
Mean Deviation |
|
|
|||
|
B3LYP |
11.4 |
2.4 |
7.9 |
|
mPW1PW91 |
-8.7 |
-0.2 |
-3.6 |
|
MPWB1K |
-9.8 |
-3.6 |
-6.2 |
|
M05-2X//B3LYP |
-6.4 |
-1.6 |
-4.1 |
|
M06-2X//B3LYP |
-4.4 |
-0.4 |
-2.5 |
|
SCS-MP2//B3LYP |
-3.2 |
-0.5 |
-1.9 |
|
|
|||
In order to discern where the problem originates, they next explore the changes that occur in the Diels-Alder reaction: two π bonds are transformed into one σ and one π bond and the conjugation of the diene is lost, leading to (proto)branching in the product. Reactions 1-3 are used to assess the energy consequence of converting a π bond into a σ bond, creating a protobranch, and the loss of conjugation, respectively.
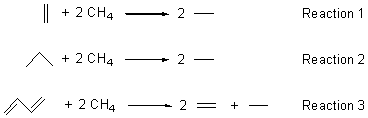
The energies of these reactions were then evaluated with the various functionals. It is only with the conversion of the π bond into a σ bond that they find a significant discrepancy between the DFT estimates and the CBS-QB3 estimate. DFT methods overestimate the energy for the π → σ exchange, by typically around 5 kcal mol-1, but it can be much worse. Relying on cancellation of errors to save the day for DFT will not work when these types of bond changes are involved. Once again, the user of DFT is severely cautioned!
References
(1) Pieniazek, S. N.; Clemente, F. R.; Houk, K. N., "Sources of Error in DFT Computations of C-C Bond Formation Thermochemistries: π → σ Transformations and Error Cancellation by DFT Methods," Angew. Chem. Int. Ed. 2008, 47, 7746-7749, DOI: 10.1002/anie.200801843

Yann responded on 01 Dec 2008 at 10:49 am #
Maybe I missed it, but the authors do not seem to address using DFT to qualitatively describe the Diels-Alder reaction. Are trends in reactivity involving various electron-donating/withdrawing group substitutions reproduced using only B3LYP, because the energy of the pi-sigma exchange is (possibly) systematic?
Marisa responded on 01 Dec 2008 at 10:57 am #
All methods in table I have empirical parameters, e.g. the scaling coefficient of SCS mp2. It is still suspicious about the accuracy if one calculate some molecule beyond the fitting database of that parameters.
Computational Organic Chemistry » Origin of DFT failure responded on 19 May 2009 at 8:09 am #
[…] with some seemingly straightforward reactions (as discussed in these previous blog posts: A, B, C, D, E, F) has become a bit clearer. Brittain and co-workers have identified the culprit.1 They […]