Dewar benzene has fascinated physical organic chemists for a long time. Just how does this open up? And why is is stable, given its large strain and the aromaticity of the ring-opened product? Most had rationalized this by recognizing that the route that takes Dewar benzene into benzene is the symmetry-forbidden disrotatory path, and the symmetry allowed conrotatory path leads to the benzene with a trans double bond.
Johnson1 discovered that the conrotatory TS is high in energy, but below the C2v structure thought to be the disrotatory TS, but is in fact a saddle point. Further, the IRC path through the conrotatory TS connects Dewar benzene to benzene, avoiding the trans-benzene (which is sometimes referred to as Möbius benzene).
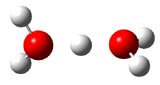
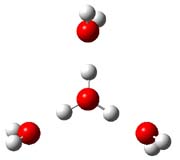
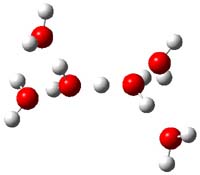
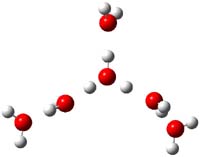
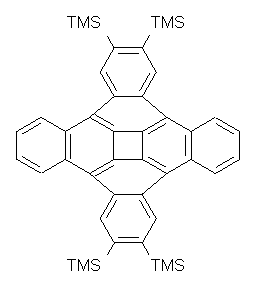
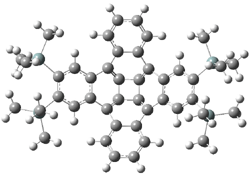
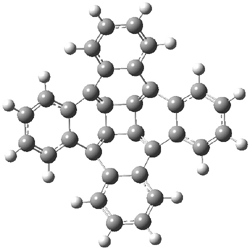
Now comes a report that the PES for opening of Dewar benzene is a bit more complicated.2 B3LYP/6-311+G** computations of 1 going to 3 identifies not only the Möat;bius benzene intermediate 2 but a transition state connecting 1 to 2 and a second transition state that connects 2 with 3. These structures are shown in Figure 1.

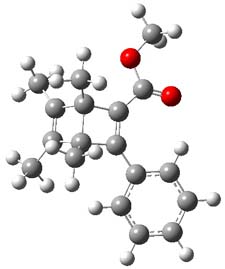 1 |
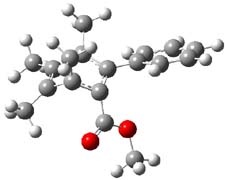 TS1 |
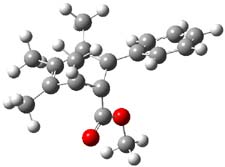 2 |
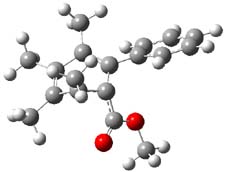 TS2 |
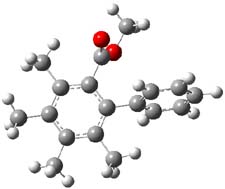
Figure 1. B3LYP/6-311+G** optimized geometries of 1-3 and the two TSs.2
The first TS is 31.0 kcal mol-1 above reactant and the Möbius benzene intermediate is only 2.1 kcal mol-1 below this TS. The second TS is 1.6 kcal mol-1 higher than the first TS, and so is rate-limiting.
The authors also examined a series of related Dewar benzenes and all have two TSs, with the second always higher than the first.
Molecular dynamics computations suggest that the Möbius benzene is likely avoided during the reaction. The fact that the two TSs are similar in energy disguised the fact that there is a second one – the energy of the first TS matches up nicely with experiments. Further MD studies would be interesting to see the interplay between the geometrically quite close intermediate and two TSs – some novel dynamics might be at work here.
References
(1) Johnson, R. P.; Daoust, K. J., "Electrocyclic Ring Opening Modes of Dewar Benzenes: Ab Initio Predictions for Möbius Benzene and trans-Dewar Benzene as New C6H6 Isomers," J. Am. Chem. Soc., 1996, 118, 7381-7385, DOI: 10.1021/ja961066q
(2) Dracinsky, M.; Castano, O.; Kotora, M.; Bour, P., "Rearrangement of Dewar Benzene Derivatives Studied by DFT," J. Org. Chem. 2010, ASAP, DOI: 10.1021/jo902065n
InChIs
1: InChI=1/C18H20O2/c1-11-12(2)18(4)15(16(19)20-5)14(17(11,18)3)13-9-7-6-8-10-13/h6-10H,1-5H3
InChIKey=LGAXKHBTTKUURV-UHFFFAOYAH
3: InChI=1/C18H20O2/c1-11-12(2)14(4)17(18(19)20-5)16(13(11)3)15-9-7-6-8-10-15/h6-10H,1-5H3
InChIKey=OUPUSCZNPIJJAH-UHFFFAOYAO