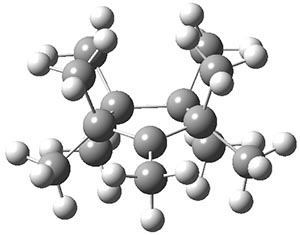
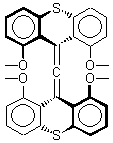
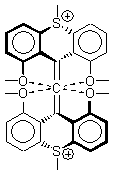
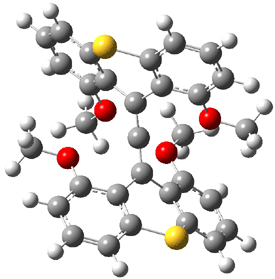
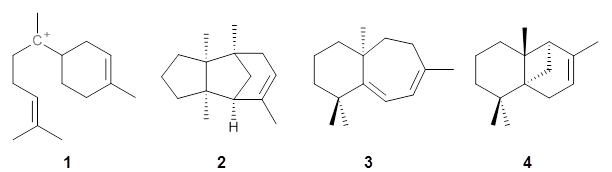
Hong and Tantillo1 report a real tour de force computational study of multiple pathways along the routes towards synthesis of a variety of sesquiterpenes. The starting point is the bisabolyl cation 1, and a variety of rearrangements, cyclizations, proton and hydride transfers are examined to convert it into such disparate products as barbatene 2, widdradiene 3, and champinene 4. The pathways are explored at mPW1PW91/6-31+G(d,p)//B3LYP/6-31+G(d,p). Some new pathways are proposed but the main points are the sheer complexity of the C15H25+ potential energy surface and the interconnections between potential intermediates.
References
(1) Hong, Y. J.; Tantillo, D. J. "Branching Out from the Bisabolyl Cation. Unifying Mechanistic Pathways to Barbatene, Bazzanene, Chamigrene, Chamipinene, Cumacrene, Cuprenene, Dunniene, Isobazzanene, Iso-γ-bisabolene, Isochamigrene, Laurene, Microbiotene, Sesquithujene, Sesquisabinene, Thujopsene, Trichodiene, and Widdradiene Sesquiterpenes," J. Am. Chem. Soc. 2014, 136, 2450-2463, DOI: 10.1021/ja4106489.
InChIs
1: InChI=1S/C15H25/c1-12(2)6-5-7-14(4)15-10-8-13(3)9-11-15/h6,8,15H,5,7,9-11H2,1-4H3/q+1
InChIKey=YKHXORRQMGBNFI-UHFFFAOYSA-N
2: InChI=1S/C15H24/c1-11-6-9-13(2)10-12(11)14(3)7-5-8-15(13,14)4/h6,12H,5,7-10H2,1-4H3/t12-,13-,14+,15-/m0/s1
InChIKey=RMKQBFUAKZOVPQ-XQLPTFJDSA-N
3: InChI=1S/C15H24/c1-12-6-7-13-14(2,3)9-5-10-15(13,4)11-8-12/h6-7H,5,8-11H2,1-4H3/t15-/m0/s1
InChIKey=SJUIWFYSWDVOEQ-HNNXBMFYSA-N
4: InChI=1S/C15H24/c1-11-6-9-15-10-12(11)14(15,4)8-5-7-13(15,2)3/h6,12H,5,7-10H2,1-4H3/t12-,14-,15-/m1/s1
InChIKey=XRDHEPAYTVHOPC-BPLDGKMQSA-N