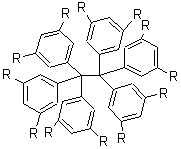
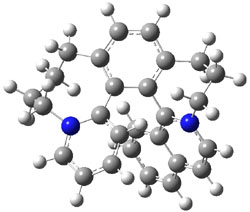
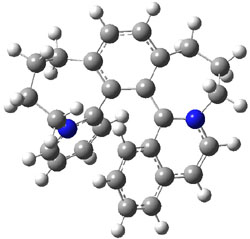
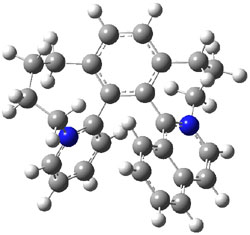
Tsuji and Nakamura have prepared the tri-p-quinodimethane 1.1 Quinodimethanes are of interest because of their possible diradical character. This new example is most interesting. It is stable as a solid in air and ambient light for 6 months, or 2 months in solution. Its ESR shows fine structure, with a spin-spin distance estimated to be 14.6 Å, very close to the distance between the terminal carbons. The ground state is a singlet, with the triplet lying 2.12 kcal mol-1 higher in energy.
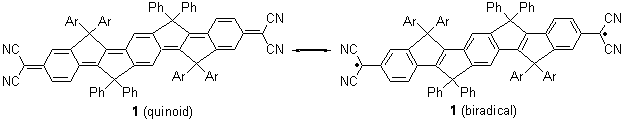
Ar = 4-octylphenyl
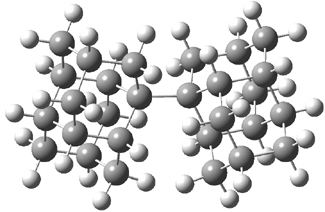
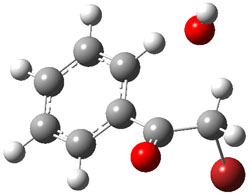
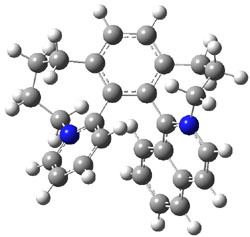
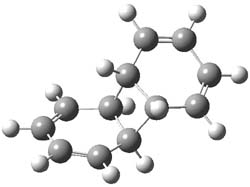
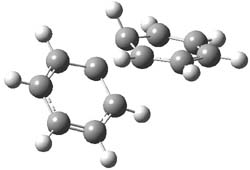
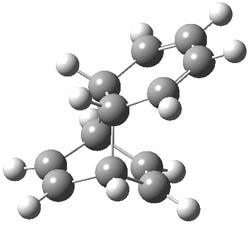
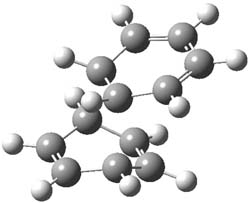
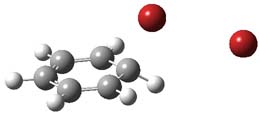
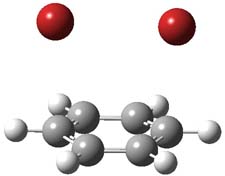
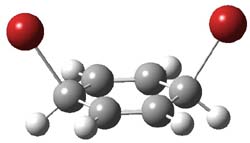
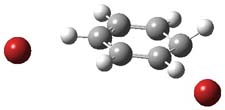
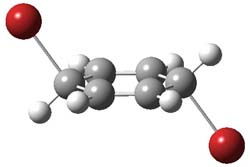
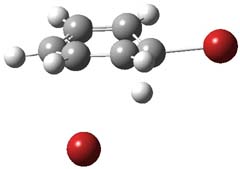
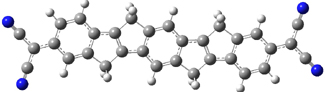
UB3LYP/6-31G** computations (lacking the aryl and phenyl sidechains) indicate a ground state singlet (with sizable spin contamination) and a gap to the triplet of 1.83 kcal mol-1. The computed geometry is shown in Figure 1.
 1 |
Figure 1. UB3LYP/6-31G** optimized geometry of 1.
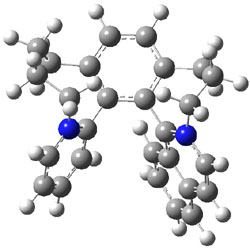
The analog having just two quinodimethane units showed no ESR signal and the computed singlet-triplet energy gap is 5.68 kcal mol-1.
It would have been interesting to have computed the NICS values for the 6-member rings – as a measure of aromatic vs. non aromatic character to further support the participation of the biradical resonance structure contribution to 1.
References
(1) Zhu, X.; Tsuji, H.; Nakabayashi, K.; Ohkoshi, S.-i.; Nakamura, E., "Air- and Heat-Stable Planar Tri-p-quinodimethane with Distinct Biradical Characteristics," J. Am. Chem. Soc., 2011, 133, 16342-16345, DOI: 10.1021/ja206060n
InChI
1: InChI=1/C112H112N4/c1-5-9-13-17-21-29-41-81-53-63-93(64-54-81)111(94-65-55-82(56-66-94)42-30-22-18-14-10-6-2)101-73-85(87(77-113)78-114)61-71-97(101)105-107(111)99-75-104-100(76-103(99)109(105,89-45-33-25-34-46-89)90-47-35-26-36-48-90)108-106(110(104,91-49-37-27-38-50-91)92-51-39-28-40-52-92)98-72-62-86(88(79-115)80-116)74-102(98)112(108,95-67-57-83(58-68-95)43-31-23-19-15-11-7-3)96-69-59-84(60-70-96)44-32-24-20-16-12-8-4/h25-28,33-40,45-76H,5-24,29-32,41-44H2,1-4H3
InChIKey=IDOIPCRGROEZHG-UHFFFAOYAD
1 (lacking aryl side chains): InChI=1/C32H16N4/c33-13-23(14-34)17-1-3-25-19(5-17)9-31-27-8-22-12-30-26-4-2-18(24(15-35)16-36)6-20(26)10-32(30)28(22)7-21(27)11-29(25)31/h1-8H,9-12H2
InChIKey=JRLKUOJPPWAWTD-UHFFFAOYAN