Nobilisitine A was isolated by Evidente and coworkers, who proposed the structure 1.1 Banwell and co-workers then synthesized the enantiomer of 1, but its NMR did not correspond to that of reported for Nobilisitine A.; the largest differences are 4.7 ppm for the 13C NMR and 0.79 ppm for the 1H NMR.2
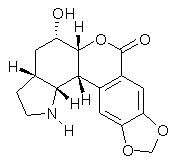
1
Lodewyk and Tantillo3 examined seven diastereomers of 1, all of which have a cis fusion between the saturated 5 and six-member rings (rings C and D). Low energy conformations were computed for each of these diasteromers at B3LYP/6-31+G(d,p). NMR shielding constants were then computed in solvent (using a continuum approach) at mPW1PW91/6-311+G(2d,p). A Boltzmann weighting of the shielding contants was then computed, and these shifts were then scaled as described by Jain, Bally and Rablen4 (discussed in this post). The computed NMR shifts for 1 were compared with the experimental values, and the mean deviations for the 13C and 1H svalues is 1.2 and 0.13 ppm, respectively. (The largest outlier is 3.4 ppm for 13C and 0.31 for 1H shifts.) Comparison was then made between the computed shifts of the seven diasteomers and the reported spectrum of Nobilisitine A, and the lowest mean deviations (1.4 ppm for 13C and 0.21 ppm for 1H) is for structure 2. However, the agreement is not substantially better than for a couple of the other diasteomers.
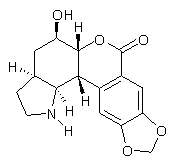
2
They next employed the DP4 analysis developed by Smith and Goodman5 for just such a situation – where you have an experimental spectrum and a number of potential diastereomeric structures. (See this post for a discussion of the DP4 method.)The DP4 analysis suggests that 2 is the correct structure with a probability of 99.8%.
Banwell has now synthesized the compound with structure 2 and its NMR matches that of the original natural product.6 Thus Nobilisitine A has the structure 2.
References
(1) Evidente, A.; Abou-Donia, A. H.; Darwish, F. A.; Amer, M. E.; Kassem, F. F.; Hammoda, H. A. m.; Motta, A., "Nobilisitine A and B, two masanane-type alkaloids from Clivia nobilis," Phytochemistry, 1999, 51, 1151-1155, DOI: 10.1016/S0031-9422(98)00714-6.
(2) Schwartz, B. D.; Jones, M. T.; Banwell, M. G.; Cade, I. A., "Synthesis of the Enantiomer of the Structure Assigned to the Natural Product Nobilisitine A," Org. Lett., 2010, 12, 5210-5213, DOI: 10.1021/ol102249q
(3) Lodewyk, M. W.; Tantillo, D. J., "Prediction of the Structure of Nobilisitine A Using Computed NMR Chemical Shifts," J. Nat. Prod., 2011, 74, 1339-1343, DOI: 10.1021/np2000446
(4) Jain, R.; Bally, T.; Rablen, P. R., "Calculating Accurate Proton Chemical Shifts of Organic Molecules with Density Functional Methods and Modest Basis Sets," J. Org. Chem., 2009, DOI: 10.1021/jo900482q.
(5) Smith, S. G.; Goodman, J. M., "Assigning Stereochemistry to Single Diastereoisomers by GIAO NMR Calculation: The DP4 Probability," J. Am. Chem. Soc., 2010, 132, 12946-12959, DOI: 10.1021/ja105035r
(6) Schwartz, B. D.; White, L. V.; Banwell, M. G.; Willis, A. C., "Structure of the Lycorinine Alkaloid Nobilisitine A," J. Org. Chem., 2011, ASAP, DOI: 10.1021/jo2016899
InChIs
2: InChI=1/C17H19NO4/c19-12-3-8-1-2-18-17(8)16-10-6-15-14(21-7-22-15)5-9(10)13(20)4-11(12)16/h5-6,8,11-12,16-19H,1-4,7H2/t8-,11-,12-,16-,17-/m0/s1
InChIKey=JISHLXUXALHAET-PUYTVRRYBF

Henry Rzepa responded on 22 Nov 2011 at 4:17 am #
I was intrigued by the observation in 10.1021/jo2016899 that they think they synthesised the enantiomer of nobilisitine. They cannot be sure however, because no chiroptical properties of the original natural product were reported! So absolute closure on this problem is not quite there yet. Thus the structure Steve draws above is (probably) NOT natural nobilisitine A, but its enantiomer (+ N-Me rather than N-H, and + a final aromatising double bond for the benzene ring to be pedantic).
I also remark that in 10.1021/jo2016899, the structure is shown rotated with respect to Steve’s representation, and the stereochemical wedges/hashes are quite different! This makes for an interesting exercise in perception to ensure that the two representations are identical (which they are). I am reminded of this in the SMILES notation, and the need to canoncalize it into a unique form in order to judge whether two SMILES strings actually represent the same molecule or not. It is interesting that there seems to be no easy way of canonicalisation of stereochemical representations if the 3D coordinates are not provided (one can always work out the CIP representation, but there are five stereo centres here, and that would take a minute or two).
Henry Rzepa responded on 01 Dec 2011 at 4:51 am #
The title of this recent article (DOI: 10.1016/j.bmc.2011.06.011) Survey of marine natural product structure revisions: A synergy of spectroscopy and chemical synthesis is both frightening (in terms of the number of mis-assignments) and encouraging (in terms of the use of computational chemistry).
I would love someone to do the same for the assignment of absolute configurations. I gave a talk on this topic recently, and from my experience, of 10 systems we looked at, 4 were in error (based it has to be said on computations).
Steven Bachrach responded on 01 Dec 2011 at 8:51 am #
Thanks to henry for finally clubbing me over the head hard enough to recognize the error in the structures of 1 and 2. These are now corrected, as is the InChI.