Can a remote substituent affect the singlet-triplet spin state of a carbene? Somewhat surprisingly, the answer is yes. Sheridan has synthesized and characterized the meta and para methoxy-substituted phenyltrifluoromethyl)carbenes 1 and 2.1 The UV-Vis spectrum of 1 is consistent with a triplet as its EPR and reactivity with oxygen. However, the para isomer 2 gave no EPR signal and failed to react with oxygen or hydrogen, suggestive of a singlet.
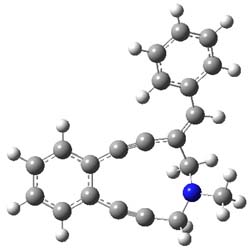
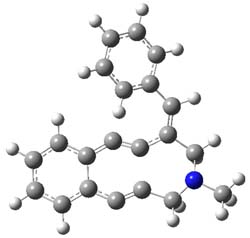
The conformations of 1 and 2 were optimized at B3LYP/6-31+G(d,p) and the lowest energy
singlet and triplet conformers are shown in Figure 1. The experimental spectral features of 1 match up best with the computed features of the triplet, and the same is true for the singlet of 2.
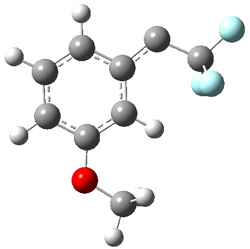 1singlet |
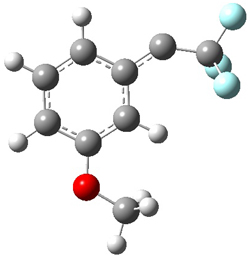 1triplet |
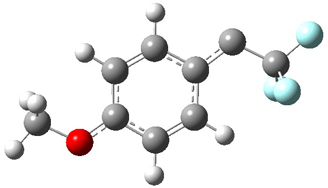 2singlet |
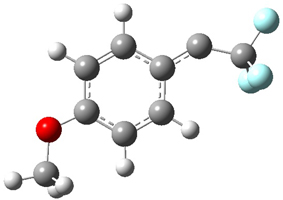 2triplet |
Figure 1. B3LYP/6-31+G(d,p) optimized geometries of 1 and 2.
The triplet of 1 is estimated to be about 4 kcal mol-1 below that of the singlet – larger than the general overestimation of the stability of triplets that beleaguer B3LYP. For 2, B3LYP predicts a singlet ground state.
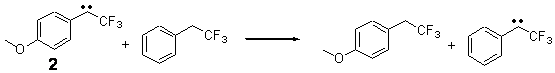
The isodesmic reactions 1 and 2 help understand the strong substituent effect. For 1, the meta substituent destabilizes both the singlet and triplet by a small amount. For 2, the para methoxy group stabilizes the triplet slightly, but stabilizes the singlet by a large amount. This stabilization is likely the result of the contribution of a second resonance structure 2b. A large rotational barrier for both the methyl methyl and the trifluoromethyl groups supports the participation of 2b.
|
|
Rxn 1 |
|
|
Rxn 2 |
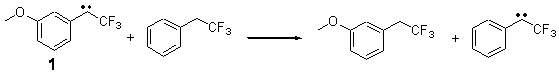
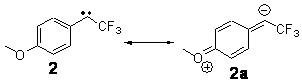
References
(1) Song, M.-G.; Sheridan, R. S., "Regiochemical Substituent Switching of Spin States in
Aryl(trifluoromethyl)carbenes," J. Am. Chem. Soc. 2011, 133, 19688-19690, DOI: 10.1021/ja209613u
InChIs
1: InChI=1/C9H7F3O/c1-13-8-4-2-3-7(5-8)6-9(10,11)12/h2-5H,1H3
InChIKey=CAHMVAJXXQQHDW-UHFFFAOYAA
2: InChI=1/C9H7F3O/c1-13-8-4-2-7(3-5-8)6-9(10,11)12/h2-5H,1H3
InChIKey