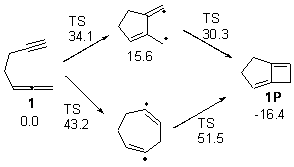
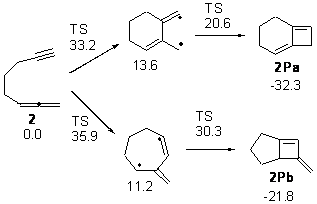
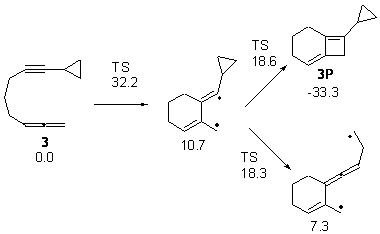
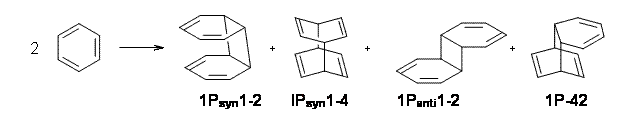
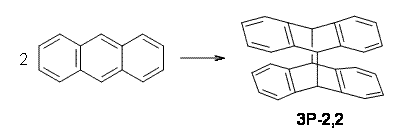
Bendikov and co-workers have examined the dimerization of linear acenes at M06-2x/6-31G(d).1 They have looked at the formal forbidden [4+4] reaction that takes, for example, 2 molecules of benzene into three possible dimer products 1Panti1-2, 1Psyn1-2, and 1Psyn1-4. The relative energies of these products increases in that order, and all three are much higher in energy than reactants; the lowest energy dimer is 1Panti1-2, lying 47.4 kcal mol-1 above two benzene molecules. Similarly, the dimerization of naphthalene is also endothermic, but the formation of the symmetric dimer of anthracene 3P-2,2’ is exothermic by 5.4 kcal mol-1. This gibes nicely with the best estimate of -9 ± 3 kcal mol-1.2 Dimerization of the higher acenes are increasingly exothermic.
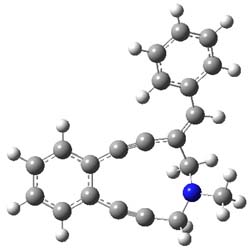
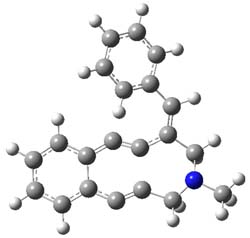
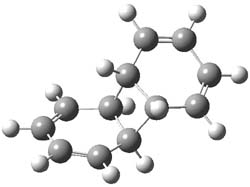
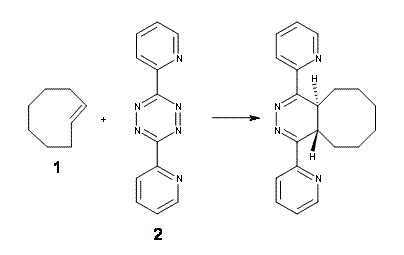
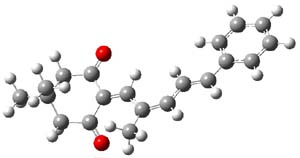
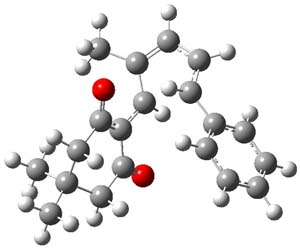
The transition state for the dimerization of benzene is concerted, though very asymmetric, as seen in Figure 1. Its energy is quite high (77.79 kcal mol-1) and so this reaction can be completely discounted. The TS for the dimerization of naphthalene is also concerted and asymmetric, but the reaction pathway for the dimerization is stepwise, with a diradical intermediate. Furthermore, the highest barrier for this stepwise reaction is 33.3 kcal mol-1. The activation energy of the back reaction (anthracene dimer to two anthrecene molecules) was measured 36.3 kcal mol-1,3 and the computed barrier of 38.7 kcal mol-1 is in nice agreement. The computed barriers for the dimerization of the higher acenes are predicted to be even lower than that of anthracene, consistent with the observation of dimers of these molecules.
|
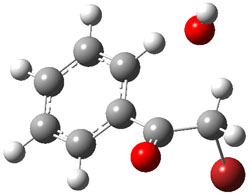
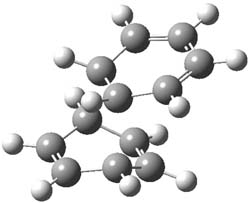
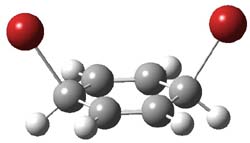
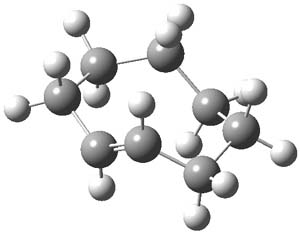
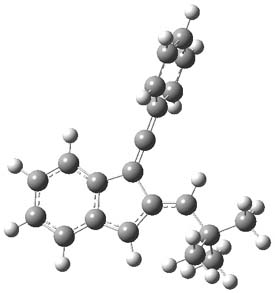
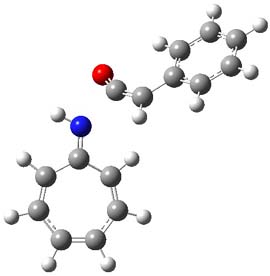
1Panti1-2
|
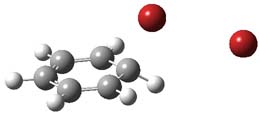
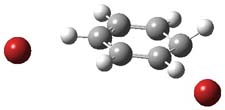
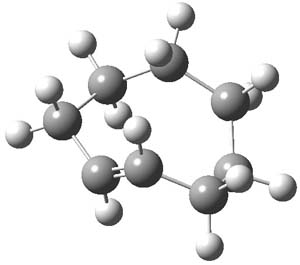
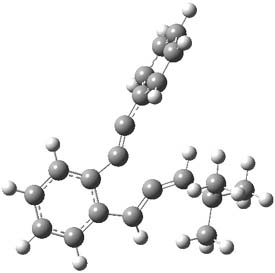
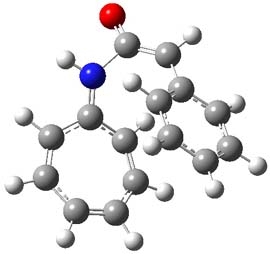
1TSanti1-2
|
|
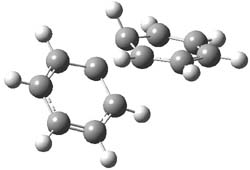
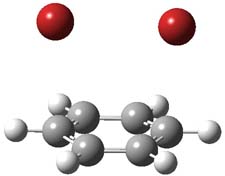
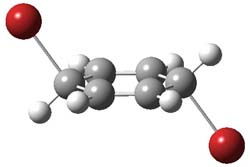
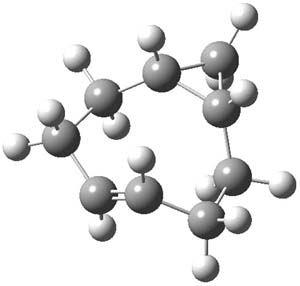
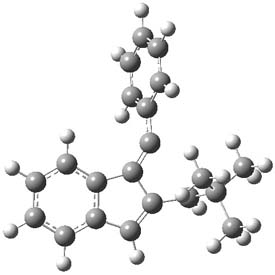
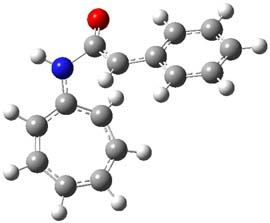
1P-42
|
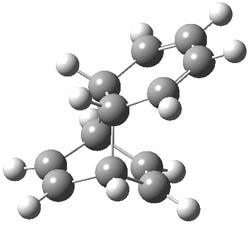
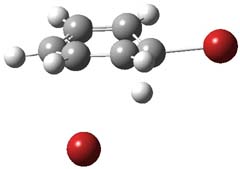
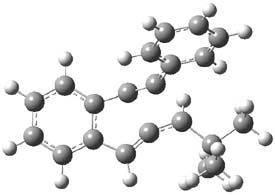
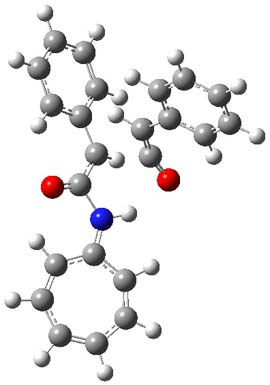
1TS-42
|
Figure 1. M06-2x /6-31G(d) optimized structures.
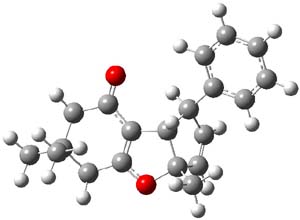
I was curious that the authors did not consider the formally allowed [4+2] dimerization, leading for example to 1P-42. So, I optimized this product and the concerted transition state leading to it. These are shown in Figure 1. The barrier through this transition state is still very large (54.1 kcal mol-1) but it is 23 kcal mol-1 lower in energy than the barrier of the [4+4] reaction! The Product of the [4+2] is also lower in energy (by 9 kcal mol-1) than 1Panti1-2. It seems to me that this type of dimerization is worth examining too – though I must say I have not as yet looked to see if anyone has explored this already.
References
(1) Zade, S. S.; Zamoshchik, N.; Reddy, A. R.; Fridman-Marueli, G.; Sheberla, D.; Bendikov,
M., "Products and Mechanism of Acene Dimerization. A Computational Study," J. Am. Chem. Soc., 2011, 133, 10803-10816, DOI: 10.1021/ja106594v
(2) Grimme, S.; Diedrich, C.; Korth, M., "The Importance of Inter- and Intramolecular van der Waals Interactions in Organic Reactions: the Dimerization of Anthracene Revisited," Angew. Chem. Int. Ed., 2006, 45, 625-629, DOI: 10.1002/anie.200502440
(3) Greene, F. D., "Problems of stereochemistry in photochemical reactions in the anthracene area," Bull. Soc. Chim. Fr., 1960, 1356-1360
InChIs
Benzene: InChI=1/C6H6/c1-2-4-6-5-3-1/h1-6H
InChIKey=UHOVQNZJYSORNB-UHFFFAOYAH
1Panti1-2: InChI=1/C12H12/c1-2-6-10-9(5-1)11-7-3-4-8-12(10)11/h1-12H
InChIKey=WMPWOGVJEXSFLI-UHFFFAOYAP
1Psyn1-2: InChI=1/C12H12/c1-2-6-10-9(5-1)11-7-3-4-8-12(10)11/h1-12H
InChIKey=WMPWOGVJEXSFLI-UHFFFAOYAP
1Psyn1-4: InChI=1/C12H12/c1-2-10-4-3-9(1)11-5-7-12(10)8-6-11/h1-12H
InChIKey=BCBHEUOKKNYIAT-UHFFFAOYAI
1P-42: InChI=1/C12H12/c1-2-4-12-10-7-5-9(6-8-10)11(12)3-1/h1-12H
InChIKey=ONVDJSCNMCYFTI-UHFFFAOYAY
Anthracene: InChI=1/C14H10/c1-2-6-12-10-14-8-4-3-7-13(14)9-11(12)5-1/h1-10H
InChIKey=MWPLVEDNUUSJAV-UHFFFAOYAK
3P-2,2’: InChI=1/C28H20/c1-2-10-18-17(9-1)25-19-11-3-4-12-20(19)26(18)28-23-15-7-5-13-21(23)27(25)22-14-6-8-16-24(22)28/h1-16,25-28H
InChIKey=JUTIJVADGQDBGY-UHFFFAOYAY