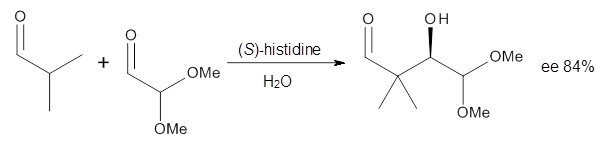
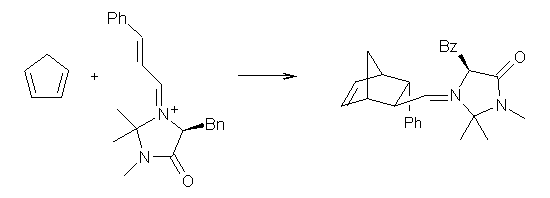
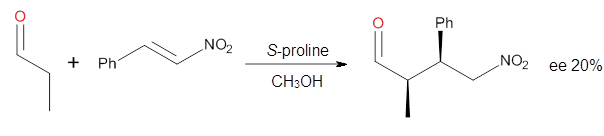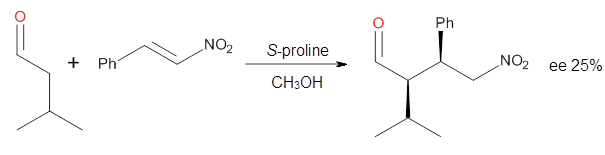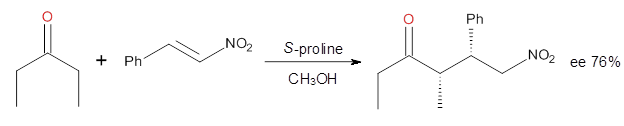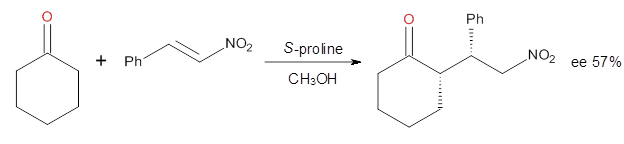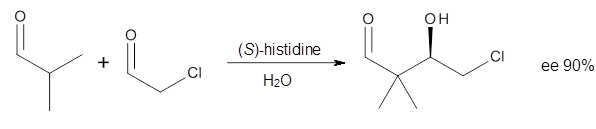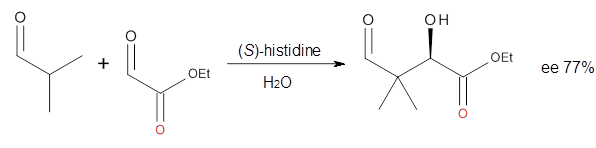Computational techniques are gaining some traction in helping to understand enantioselective organocatalysis. I talk about a few examples in Chapter 6.3 of my book. Lambert and Vetticatt have now used computations to help understand the role of the catalyst 4 in the Michael addition shown in Scheme 1.1 This reaction proceeds with 99% yield and an ee of 98%.
Scheme 1.
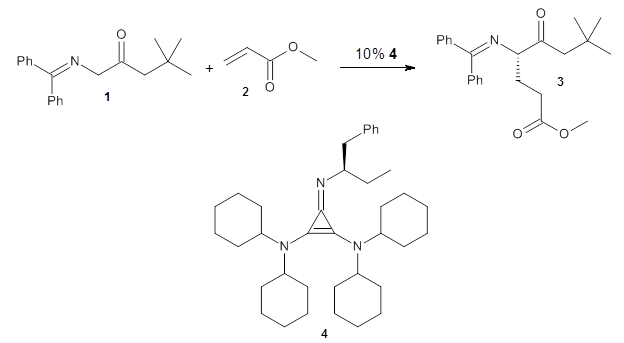
13C kinetic isotope effect studies suggest that the rate determining step is the C-C bond formation (the Michael addition step) which follows the deprotonation of the imine 1 by the catalyst 4.
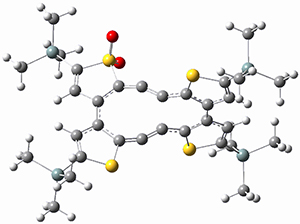
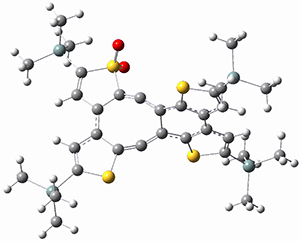
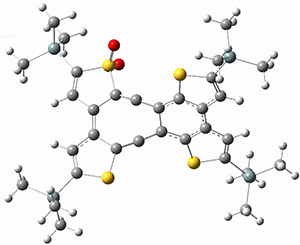
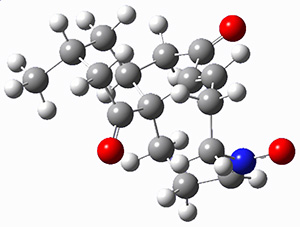
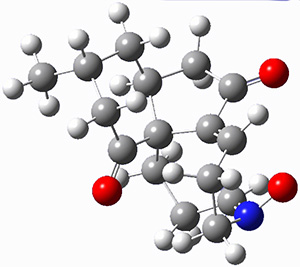
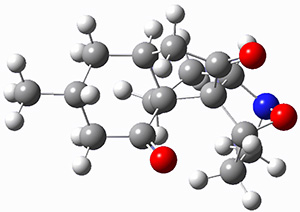
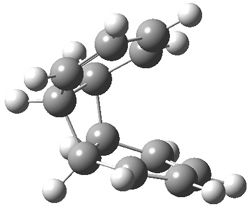
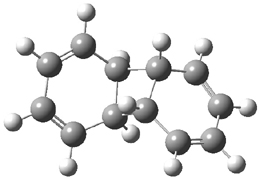
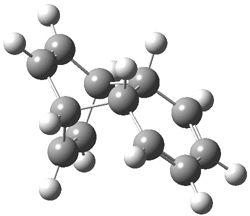
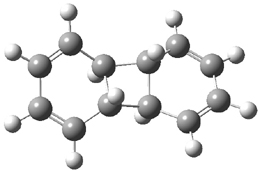
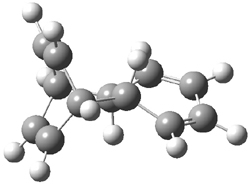
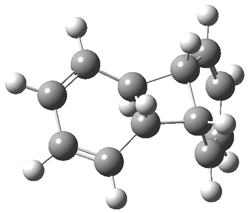
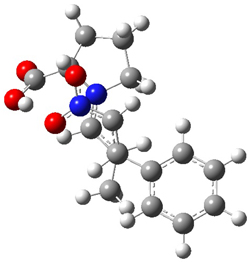
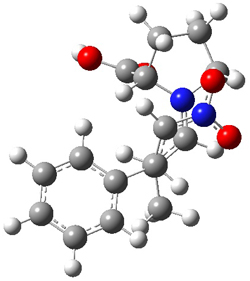
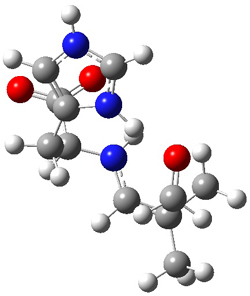
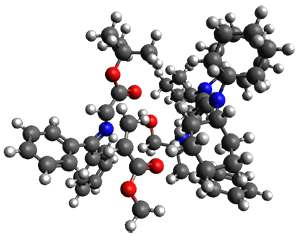
They performed ONIOM computations to search for transition states of this rate limiting step for the reaction in Scheme 1, using the full molecules. From this ONIOM search, the energies for all transition structures with 5 kcal mol-1 of the lowest energy structure were then obtained at B3LYP/6-31G*. The three lowest energy TS are shown in Figure 1. The two lowest energy structures lead to the major enantiomer, while the third lowest energy structure leads to the minor enantiomer. These energies lead to a prediction of an ee of 92%, in reasonable agreement with the experiment. The computed kinetic isotope effects are in nice agreement with experiment, supporting this step as the overall rate limiting step.
|
TSs leading to the S isomer |
|
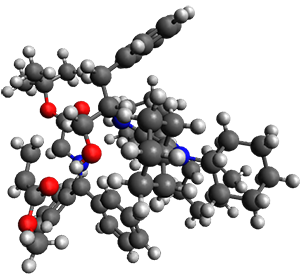
TS1 |
TS2 |
|
TS leading to the R isomer |
|
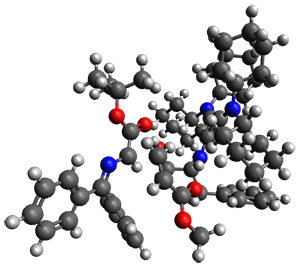
TS3 |
|
Table 1. ONIOM optimized geometries of the three lowest energy TSs. Relative energy (kcal mol-1) in parenthesis.
Analysis of what factors are important in determining the ee is complicated and ultimately the authors are unable to provide a simple explanation. They properly note that
The observation that the major enantiomer (S) is formed from two very geometrically distinct transition structures … suggests that the prediction of enantioselectivity for other reactions … will require a full consideration of all possible transition state assemblies. (emphasis mine)
I agree with this sentiment, pessimistic as it may be. Answering this type of question is likely to remain very challenging for years to come.
References
1) Bandar, J. S.; Sauer, G. S.; Wulff, W. D.; Lambert, T. H.; Vetticatt, M. J. "Transition State Analysis of Enantioselective Brønsted Base Catalysis Chiral Cyclopropenimines," J. Am. Chem. Soc. 2014, 136, 10700-10707, DOI: 10.1021/ja504532d.
InChIs
1: InChI=1S/C20H23NO/c1-20(2,3)14-18(22)15-21-19(16-10-6-4-7-11-16)17-12-8-5-9-13-17/h4-13H,14-15H2,1-3H3
InChIKey=UZCWUGCTNCNJHI-UHFFFAOYSA-N
2: InChI=1S/C4H6O2/c1-3-4(5)6-2/h3H,1H2,2H3
InChIKey=BAPJBEWLBFYGME-UHFFFAOYSA-N
3: InChI=1S/C24H29NO3/c1-24(2,3)17-21(26)20(15-16-22(27)28-4)25-23(18-11-7-5-8-12-18)19-13-9-6-10-14-19/h5-14,20H,15-17H2,1-4H3/t20-/m0/s1
InChIKey=KTASCPHNNZODSX-FQEVSTJZSA-N
4: InChI=1S/C37H57N3/c1-2-30(28-29-18-8-3-9-19-29)38-35-36(39(31-20-10-4-11-21-31)32-22-12-5-13-23-32)37(35)40(33-24-14-6-15-25-33)34-26-16-7-17-27-34/h3,8-9,18-19,30-34H,2,4-7,10-17,20-28H2,1H3/t30-/m1/s1
InChIKey=GEHSIGXXLTVFFG-SSEXGKCCSA-N