Here is a nice example of an interesting synthesis, mechanistic explication using computation (with a bit of an unanswered question), and corroboration of the stereochemistry of the product using computed NMR shifts. Gil and Mischne1 reacted dimedone 1 with dienal 2 under Knoevenagel conditions to give, presumably, 3. But 3 is not recovered, rather the tricycle 4 is observed.
There are four stereoisomers that can be made (4a-d). Computed 13C chemical shifts at OPBE/pcS-1 (this is a basis set suggested for computing chemical shifts2) for these four isomers were then compared with the experimental values. The smallest root mean squared error is found for 4d. Better still, is that these authors utilized the DP4 method of Goodman3 (see this post), which finds that 4d agrees with the experiment with 100% probability!
Lastly, the mechanism for the conversion of 3 to 4 was examined at M06/6-31+G**. The optimized geometries of the starting material, transition state, and product are shown in Figure 1. The free energy barrier is a modest 14.5 kcal mol-1. The TS indicates a conrotatory 4πe– electrocyclization. The formation of the C-O bond lags far behind in the TS. They could not identify a second transition state. It would probably be worth examining whether the product of this 4πe– electrocyclization could be located, perhaps with an IRC starting from the transition state. Does this TS really connect 3 to 4?
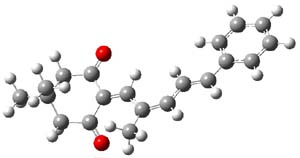 3 |
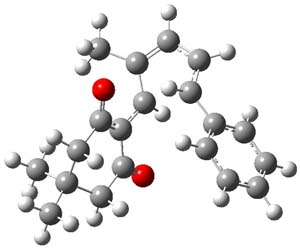 TS |
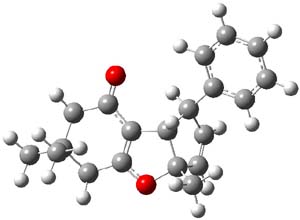 4 |
|
Figure 1. M06/6-31+G** optimized geometries of 3 and 4 and the transition state connecting them.
References
(1) Riveira, M. J.; Gayathri, C.; Navarro-Vazquez, A.; Tsarevsky, N. V.; Gil, R. R.; Mischne, M. P., "Unprecedented stereoselective synthesis of cyclopenta[b]benzofuran derivatives and their characterisation assisted by aligned media NMR and 13C chemical shift ab initio predictions," Org. Biomol. Chem., 2011, 9, 3170-3175, DOI: 10.1039/C1OB05109A
(2) Jensen, F., "Basis Set Convergence of Nuclear Magnetic Shielding Constants Calculated by Density Functional Methods," J. Chem. Theory Comput., 2008, 4, 719-727, DOI: 10.1021/ct800013z
(3) Smith, S. G.; Goodman, J. M., "Assigning Stereochemistry to Single Diastereoisomers by GIAO NMR Calculation: The DP4 Probability," J. Am. Chem. Soc., 2010, 132, 12946-12959, DOI: 10.1021/ja105035r
InChIs
1: InChI=1/C8H12O2/c1-8(2)4-6(9)3-7(10)5-8/h3-5H2,1-2H3
InChIKey=BADXJIPKFRBFOT-UHFFFAOYAX
2: InChI=1/C12H12O/c1-11(10-13)6-5-9-12-7-3-2-4-8-12/h2-10H,1H3/b9-5+,11-6+
InChIKey=VFBDYWDOVMUDEB-MPEOSAONBY
3: InChI=1/C20H22O2/c1-15(8-7-11-16-9-5-4-6-10-16)12-17-18(21)13-20(2,3)14-19(17)22/h4-12H,13-14H2,1-3H3/b11-7+,15-8+
InChIKey=IBEGRISKTNRVOU-YQQAFNMCBC
4d: InChI=1/C20H22O2/c1-19(2)11-15(21)17-16(12-19)22-20(3)10-9-14(18(17)20)13-7-5-4-6-8-13/h4-10,14,18H,11-12H2,1-3H3/t14-,18+,20+/m1/s1
InChIKey=VEGSTZFBNRXEAX-WNYOCNMUBZ

Rob Paton responded on 27 Aug 2011 at 8:34 am #
Taking a closer look at the imaginary frequency of the TS, it doesn’t look much like a 4pi-electrocyclization, rather a vinylogous Michael addition. In this picture, it is a diene that attacks an enone forming the 5m-carbocycle, in the process forming a tertiary carbocation that is then trapped by the enolate oxygen to form the cyclic ether. The formation of this second ring would therefore be facile so I’m not surprised the authors do not locate a second TS. I haven’t followed the IRC though.
Second observation: reusing Cartesian coordinates is lot less painful when taken from Jmol figures (like those found on this blog) than from SI PDFs.
Steven Bachrach responded on 27 Aug 2011 at 9:44 am #
Good point about the mechanism perhaps being a Michael addition – that does explain the lack of a second TS.
And I completely agree with your point about reusing coordinates – this can often be quite painful! But I am in general passified if they have at least included the coordinates at all!
Armando Navarro responded on 05 Sep 2011 at 4:58 am #
Following the IRC stops in a cyclopentenyl zwitterionic point with all-positive vibrational frequencies. However, scanning the C—O coordinate from this point does not show any evidence of a TS, so it can be better classified as an inflexion point rather than a detectable intermediate in the surface. The potential surface is very similar to that studied by Silva López et al ( DOI: 10.1021/jo0603274 ). See the Figure 6 in this paper for a nice graphical representation of the potential surface of this kind of reactions