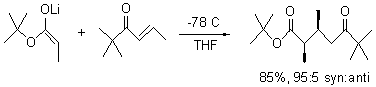
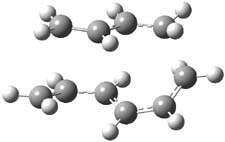
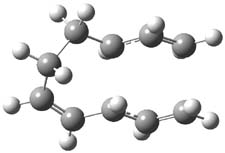
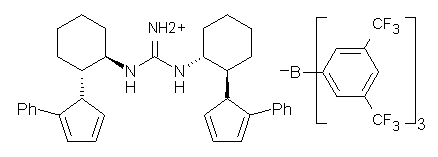
Jacobsen reports another interesting example of organocatalysis, here using a chiral guanadinium salt to catalyze the enantioselective Claisen rearrangement.1 As an example, Reaction 1 proceeds in 6 days at 30 °C to give 81% yield with an ee of 84%. The system is also diastereoselective, so that Reaction 2, run for 6 days at 40 °C, gives an 82% yield with a diastereomeric ratio of 16:1 and an ee of 81%.
|
|
Reaction 1 |
|
|
Reaction 2 |
|
|
|


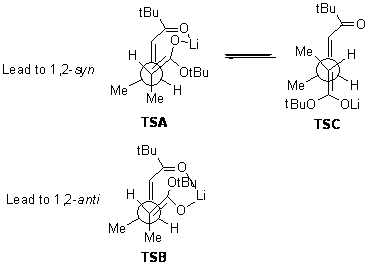
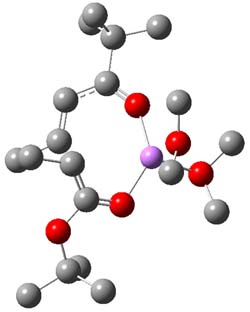
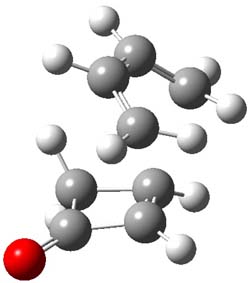
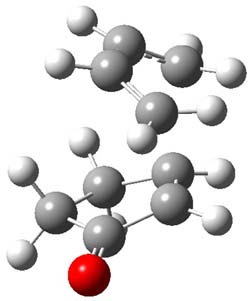
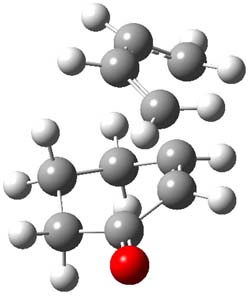
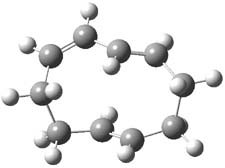
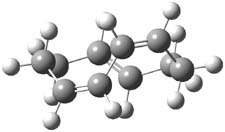
B3LYP/6-31G(d,p) computations provide some insight. The uncatalyzed reaction of 1 to give 2 is predicted to be exothermic by 16.1 kcal mol-1, with an activation energy of 25.9 kcal mol-1. Using N,N’-dimethylguanidnium as a model for the catalyst (and with no counter anion and no treatment of solvent – hexanes in this case), they find a complexation energy of almost 27 kcal mol-1 for forming 3. 3 exhibits (See Figure 1) three hydrogen bond-like interactions – one N-H bifurcates to interact with the carbonyl oxygen and (a very long interaction) to the other oxygen. The product complex 4 also shows three hydrogen bond-like interactions, with an overall exothermicity of -14.7 kcal mol-1. The complexed transition state 5 has two normal length hydrogen bonds, with an activation energy above 3 of 20.6 kcal mol-1. Thus the complex lowers the barrier by about 5 kcal mol-1, indicating the catalytic effect. They have not however addressed the enantioselectivity.
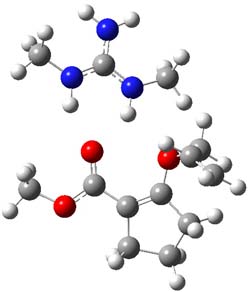 3 |
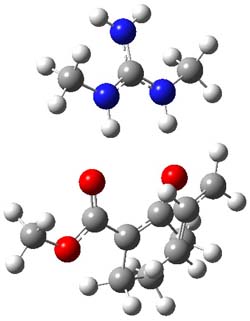 5 |
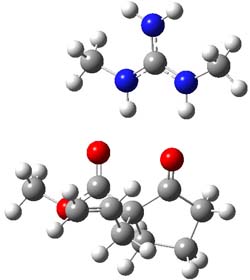 4 |
|
Figure 1. B3LYP/6-31G(d,p) optimized geometries of 3-5.
References
(1) Uyeda, C.; Rötheli, A. R.; Jacobsen, E. N., "Catalytic Enantioselective Claisen Rearrangements of O-Allyl β-Ketoesters," Angew. Chem. Int. Ed., 2010, 49, 9753–9756, DOI: 10.1002/anie.201005183
InChIs
1: InChI=1/C10H14O3/c1-3-7-13-9-6-4-5-8(9)10(11)12-2/h3H,1,4-7H2,2H3
InChIKey=NASFSRKGDOBHIX-UHFFFAOYAC
2: InChI=1/C10H14O3/c1-3-6-10(9(12)13-2)7-4-5-8(10)11/h3H,1,4-7H2,2H3/t10-/m0/s1
InChIKey=QXKXLNGEBVMWLH-JTQLQIEIBT