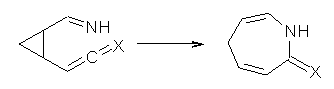
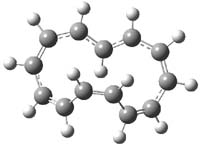
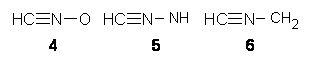Grinberg and colleagues have published a combination of VCD and computation to understand the racemization of imidazoline 1 when exposed to base.1 Experimental VCD performed at various temperatures indicates first-order kinetics with a barrier of about 24 kcal mol-1.
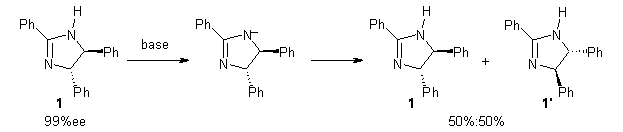
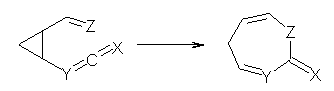
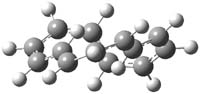
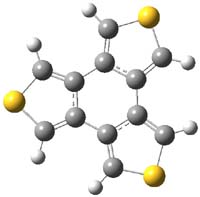
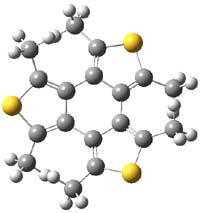
The mechanism for this racemization was proposed and supported with B3LYP/6-31G(d) computations. The anion of 1 can undergo a disrotatory ring opening to form 2, passing through TS1 with a barrier of about 21 kcal mol-1. Since 2 is chiral with the phenyl groups oriented in non-equivalent positions, ring closure of 2 will go back to 1 and not on to its racemate. In order to racemize, 2 must convert to 3, which can invert to 3’ and then on to 1’. While the barrier for ring opening is likely to be rate limiting, and it does match up reasonably well with the experimental value, the authors have not optimized the transition state that take 2 into 3 or the TS that interconverts 3 with 3’. It’s the former TS that may be pretty large as it requires disruption of the conjugation. Unfortunately, not only have the authors not computed these other TSs, the supplementary materials include only the optimized structure of 1 and not TS1, 2, or 3!

The authors do note that the ring opening is facilitated by the phenyl group on the chiral carbons of 1. They replaced the phenyls with cyclohexyl or cyclohexenyl groups and racemization is no longer observed. Strangely, the authors include in the supporting materials the optimized structures of these variants, but not the TSs for ring opening. Thus, the confirming evidence of a very high barrier for ring opening that would really nail down the mechanism is missing!
References
1) Ma, S.; Busacca, C. A.; Fandrick, K. R.; Bartholomeyzik, T.; Haddad, N.; Shen, S.; Lee, H.; Saha, A.; Yee, N.; Senanayake, C.; Grinberg, N., "Directly Probing the Racemization of Imidazolines by Vibrational Circular Dichroism: Kinetics and Mechanism," Org. Lett., 2010, 12, 2782–2785, DOI: 10.1021/ol100734t
InChIs
1: InChI=1/C21H18N2/c1-4-10-16(11-5-1)19-20(17-12-6-2-7-13-17)23-21(22-19)18-14-8-3-9-15-18/h1-15,19-20H,(H,22,23)/t19-,20-/m0/s1/f/h22H
InChIKey=UCCFUHZMGXEALP-RLNNBPQHDR