I have discussed Möbius aromatic systems in the book and in the blog. A new Möbius aromatic platform has been synthesized, where the porphyrin π-system is appropriately twisted. Osuka has prepared the hexaphyrins 1 and 2.1 These possess a double-twist structure, and with its 28 π-electrons 1 should be antiaromatic and 2, having 26 π-electrons should be aromatic.
|
|
|
|
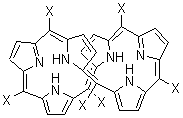
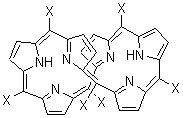
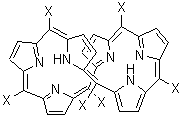
In fact, the x-ray structure of 1 displays significant bond alternation and the NH protons (in the interior of the molecule) have chemical shift far downfield (δ 14.95 and 12.35 ppm) – all consistent with antiaromatic character. On the other hand¸ while 2 exhibits little bond alternation, the NH protons are seen at 11.1 ppm, too far downfield for the interior positions of an aromatic compound!
Rzepa2 has computed 1 and 2 at MPW1PW91/6-31G(d,p) for X=H and CF3; the latter matches the experimentally prepared compounds. (Rzepa supplies very nice web-enabled access to his results through the supporting materials, and so I do not repeat his structures here. Please also see comments to this post.) As expected, both optimized structures of 1(X=H or X=CF3) shows distinct bond localization and positive NICS values. The chemical shifts of the NH protons are far downfield, and in reasonable agreement with the experimental shifts. The optimized structures of 2 display bond delocalization and negative NICS values, indicative of aromaticity, as do the NH chemical shifts of 5.2 ppm (X=CF3) or 3.8 ppm (X=H). These chemical shifts differ from the experiment. Rzepa locates a second less stable conformation 3, but its NH chemical shifts are at 10.9 and 10.1 ppm, in reasonable agreement with experiment. So, he concludes that 1 is antiaromatic and 2 is aromatic and both have a double-twist Möbius topology.
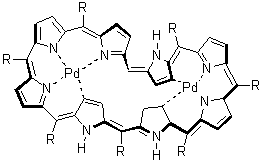
Tanaka, et al have reported the structure of the octaphyrin held in place by a complexed
metal, such as 4.3 A number of analogues have been prepared and their x-ray structure shows the single twist needed for Möbius topology. The NMR spectra are consistent with an aromatic system. And relevant to this blog, B3LYP/6-31G(d) (SDD for the heavy metals) NICS computations reveals a large negative value, -14.6 ppm for 5.
|
|
References
(1) Shimizu, S.; Aratani, N.; Osuka, A., "meso-Trifluoromethyl-Substituted
Expanded Porphyrins," Chem. Eur. J., 2006, 12, 4909-4918, DOI: 10.1002/chem.200600158
(2) Rzepa, H. S., "Lemniscular Hexaphyrins as Examples of Aromatic and Antiaromatic
Double-Twist Möbius Molecules," Org. Lett. 2008, DOI: 10.1021/ol703129z
(3) Tanaka, Y.; Saito, S.; Mori, S.; Aratani, N.; Shinokubo, H.; Shibata, N.; Higuchi, Y.; Yoon, Z. S.; Kim, K. S.; Noh, S. B.; Park , J. K.; Kim , D.; Osuka, A., "Metalation of Expanded Porphyrins: A Chemical Trigger Used To Produce Molecular Twisting and Möbius Aromaticity," Angew. Chem. Int. Ed., 2008, 47, 681-684, DOI: 10.1002/anie.200704407
InChIs
1(X=H): InChI=1/C30H22N6/c1-2-20-14-22-5-6-24(33-22)16-26-9-10-28(35-26)18-30-12-11-29(36-30)17-27-8-7-25(34-27)15-23-4-3-21(32-23)13-19(1)31-20/h1-18,31-32,35-36H/b19-13-,20-14-,21-13+,22-14-,23-15-,24-16-,25-15-,26-16-,27-17-,28-18+,29-17-,30-18-
InChIKey: LZQBSNZLFFXDHG-FXYKPCLQBX
1(X=CF3): InChIKey: XMLQOLTZAIQLSA-UFROFZBYBD
2(X=H): InChI=1/C30H20N6/c1-2-20-14-22-5-6-24(33-22)16-26-9-10-28(35-26)18-30-12-11-29(36-30)17-27-8-7-25(34-27)15-23-4-3-21(32-23)13-19(1)31-20/h1-18,31,36H/b19-13-,20-14-,21-13-,22-14-,23-15-,24-16+,25-15+,26-16-,27-17-,28-18-,29-17-,30-18-
InChIKey: MMFGRQLRZMFRGC-NJRWMLEBBC
2(X=CF3): InChIKey: FFCNJJQCGZCFGC-FJVZODTDBS

ChemSpiderMan responded on 21 Feb 2008 at 8:29 am #
The link to Rzepa’s supporting materials doesn’t appear to be active Steve.
sbachrach responded on 21 Feb 2008 at 9:37 am #
The link is now fixed. However, it turns out that this material is actually not supporting material but a web-enhanced object and so you will need to have subscription access to get to it. Rzepa has also placed the output files on a digital repository at Imperial college. So here are the DOI links to those files: 1(X=H): 10042/to-426, 1(X=CF3): 10042/to-427, 2(X=H): 10042/to-428, 2(X=CF3): 10042/to-428, 3(X=H): 10042/to-447, 3(X=CF3): 10042/to-444.
Henry Rzepa responded on 26 Feb 2008 at 2:14 am #
Jut a typo; where you write “The optimized structures of 2 display bond localization and negative NICS values, indicative of aromaticity” I think you meant:
“The optimized structures of 2 display bond delocalization and negative NICS values, indicative of aromaticity”
As for the web-enhanced object, I should emphasize that the copyright for this object is NOT owned exclusively by the ACS, who agreed not to claim in since it comprised data (which is not copyrightable). This is something of a first for the ACS, who traditionally have claimed such copyright for all supporting information submitted by authors.
Henry Rzepa responded on 26 Feb 2008 at 2:19 am #
Might I flag for discussion the use of a digital repository for archiving the full data for a calculation. Unless I am wrong, this is the first instance of such use in chemistry. There are a number of observations
1. The data is date stamped. Thus it might be useful when issues of priority arise. The date of course does not establish when the calculation was actually done, merely when it was first exposed to public in the repository.
2. Each molecular entry contains the InChI and InChI key. Hopefully, they agree with the ones Steve quotes in this blog!
3. Our repository also accepts inputs from NMR spectra and crystallography, so it is also of interest to experimental chemists
4. An article submitted to J. Chem. Inf. Mod. describes the whole process of creating such a repository. We urge all computational chemists who publish to operate such a repository!
Henry Rzepa responded on 26 Feb 2008 at 2:27 am #
Sorry, another typo. Compound 2 is generally counted as having [26] not [30] π-electrons. How to count the electrons in such phyrins is an interesting exercise, since not all of the π electrons are actually counted!
sbachrach responded on 26 Feb 2008 at 7:51 am #
Thanks Henry – I have corrected the two typos.
Your means for digital deposition is quite ingenious – very easy to use and all pertinent data is nicely collected. My only real complaint is not with the copyright (where there is really something to debate) but the fact that ACS treats this as a part of the article and not as supplementary information. Supplementary information is made available for free to all while the web-enhanced objects, treated as a component of the article, are available only to subscribers or by purchase. The novel way that Henry has used the web-enhanced object here blurs the distinction entirely – it is both supplementary material and an integral part of the article – and ACS (and other publishers) will need to rethink how this data/intellectual creation is treated!
Henry Rzepa responded on 26 Feb 2008 at 8:23 am #
Steve raises the interesting issue of whether the data should be part of the main article (where it is inaccessible to non-subscribers) or supplemental (where it is accessible). However, the latter is often regarded very much as a second-rate citizen in the publishing world, it is often not indexed, and in the past, too many TIFF files (and other almost unreadable objects) have meant it is viewed as both unimportant, and low quality (no proper validation processes are ever applied to it, excepting possibly CIF files).
Steve and I are both veterans of a brave experiment to indeed blur the distinction between a primary publication and supporting data, with the Internet Journal of Chemistry. That was probably ahead of its time! What I would suggest now is that with publishers slowly bringing data into mainstream articles, and with readers hopefully appreciating the benefits this brings, that gradually the evolution of the journal into a proper Web 3.0 environment will start to accelerate.
Mind you, when I mentioned this concept to a colleague (who will be nameless) he was horrified. It meant that others could rapidly reproduce reported calculations, and hence steal a march on the original authors. In effect, a lot of people were happy with information by obscurity; it buys them time to get the credit they think they deserve. So the concepts outlined here are apparently not for these guys!
Henry Rzepa responded on 26 Feb 2008 at 8:36 am #
A quick comment on the links Steve has put to our digital repository. He refers to them as DOI links. Whilst it is true that they are properly resolved by the full URL http://dx.doi.org/10042/to-427, they cannot and should not be referred to as a formal DOI. This is a licensed object purchased from a commercial organization, and we have not done this with our identifiers. Instead, the generic name is a handle, which the DOI organization kindly resolves for us. To avoid such confusion, but to keep the simple-to-use association with a DOI that most chemists have now become familiar with, I proposed the term COI, but my colleagues on the project had some reservations about the use of that term (the collection of files found at the repository is not a formal object in itself).
I should also clarify that whilst digital repositories themselves are probably decades old, the DSpace utility which we use is new in that it exposes the metadata for the collection in a formal, and harvestable manner (facilitating Google and other trawling), and will enable automatic aggregation in a manner not previously possible.
sbachrach responded on 26 Feb 2008 at 9:30 am #
I have become an even greater believer in supporting information while I wrote my book. I wanted to include drawings of the structures of all the molecules I present in the text in a uniform manner. This meant obtaining the optimized geometries and piping them into my molecular visualization tool (I use GaussView). But to my chagrin, many, many authors do not publish their structures in any form. The drawings in the actual paper usually do not include all of the geometrical parameters. Many authors deposit no supplemental (supporting) information. I was left to having to reoptimize geometries of many molecules.
The bottom line is that I am now a complete believer in the Open Data concept, a concept that Peter Murray-Rust has been perhaps the greatest champion.
Henry Rzepa responded on 26 Feb 2008 at 9:43 am #
Agreed Steve. It is of course not an entirely new concept; one possible start to it was in 1994, when a small group of us proposed MIME standards for exchanging data using the Internet and Web, and used this in eg Chime etc. So even by that date, its 14 years old! Its been bubbling away ever since of course; the difference is that now it has been the Trojan horse that has entered the city of the mainstream publishers!
Also, as a referee of many articles, I often ask the authors to send coordinates of calculations/molecules they report, since they do not do so when submitting. This often irritates them, but they always obey (ie they want their article published!). This can occasionally be most revealing! Indeed, I sometimes suspect that this insight the referee can get is something the authors would rather wish they did not get!
But how many times does this happen. Of 99 (999?) articles refereed, how many have/use such data! I once sent a ZIP file as part of an article to be refereed (it has to be said with no processing instructions for that file) containing about 100 coordinate files, all to be viewed with Jmol). A bad-tempered referee comment came back saying they had tried to open the archive in Microsoft Word, failed, and this simply reinforced their belief that the article was not worth publishing!
sbachrach responded on 26 Feb 2008 at 9:47 am #
As the kids say: LOL!
Henry Rzepa responded on 26 Feb 2008 at 9:56 am #
Perhaps another connected anecdote. It was this Blog where I spotted Steve’s commentary on the famous Hexacyclinol saga, and Scott Rychnovsky’s reassignment of the structure. Reading Scott’s article, it seemed to me that the methodology might be improvable, so I decided to repeat his calculations. Having access only to the ACS supporting information, I toiled for about 2 hours to get the coordinates in the Acrobat file (the manner in which the data is distributed) into Gaussview. It kept crashing! The culprit was in fact 27 significant, but erroneous white spaces. Thus a space would break up a floating point number, from 1.234 into 1. 234 (ie two numbers). i repeated this for the second molecule. Four hours later, I was up and running. It should have taken 30 seconds! I made a few modifications, and found that Scott’s errors for the 13C shifts could be reduced by a factor of two. A colleague walked into my office; I mentioned what I had done. Well, he said, I have a challenge for you! The rest of the story, if you are interested, can be see at http://dx.doi.org/10.1021/np0705918 Determined to avoid the pain I had encountered, I venture to suggest anyone wishing to repeat our analysis can now do this in 30 seconds!
Oh, you may ask where those 27 spaces came from. I asked Scott, but he is baffled. No-one knows! We could start a thread here on validation if we wished!
Computational Organic Chemistry » Hexaporphyrin that’s Möbius aromatic responded on 07 Jul 2009 at 7:55 am #
[…] produced without the need for low temperature, complexation with a metal or protonation (see this post for a discussion of their earlier work). The x-ray crystal structure shows the Möbius twist, […]