In order to obtain computed NMR chemical shifts, one computes the isotropic magnetic shielding tensor and subtracts this value from that computed for a reference (or standard) compound. Typically, one uses TMS as the standard. Sarotti and Pellegrinet have questioned whether this is a reasonable approach.1 Since computational methods vary in quality with methodology, basis set, geometry – one might wonder if the use of a single standard for all computed chemical shifts is the best approach.
They computed the 13C chemical shielding tensor for 50 organic compounds possessing a wide variety of functional groups and rings – a few examples are given below. They also computed the 13C chemical shielding tensor for 11 different simple organic compounds that might be used as NMR references (like TMS, benzene, methanol, and chloroform).
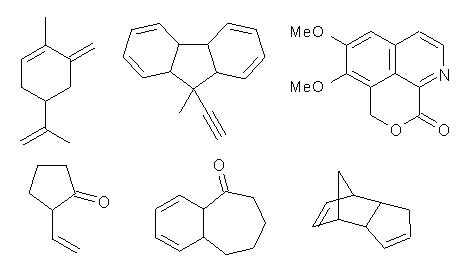
By comparing the computed chemical shifts obtained using the different references and then matching them with experiment, they propose a multi-reference method. For sp3 carbon atoms they propose using methanol as the reference, and for sp2 and sp carbons using benzene as the reference. With chemical shifts computed at mPW1PW91/6-311+G(2d,p)//B3LYP/6-31G(d) using the multi-reference model , the average mean difference from experiment is 2.1 ppm, less than half that found when TMS alone is used. The average RMS deviation of 4.6ppm is about half that when TMS is used as the sole standard.
Though the authors mention the solvent effect on chemical shifts, it is surprising that they did not include solvent in their calculations, especially since they are comparing to experimental chemical shifts in deuterochloroform. Nonetheless, I think this is a nice idea and further exploration of this concept (multi-reference fitting) is worth further pursuits.
References
(1) Sarotti, A. M.; Pellegrinet, S. C., "A Multi-standard Approach for GIAO 13C NMR Calculations," J. Org. Chem., 2009, 74, 7254-7260, DOI: 10.1021/jo901234h

Fabio Costa responded on 26 Aug 2010 at 9:35 am #
Indeed It is worth to check out! Recently we published another interesting paper focused on 13C NMR chemical shifts calculations. We claim to our scaling factor protocol high accuracy and low computational cost. Although behaviour effects were not considered, it also were able achieved great experimental data reproduction.
Fabio Costa responded on 26 Aug 2010 at 9:37 am #
Indeed It is worth to check out! Recently we published another interesting paper focused on 13C NMR chemical shifts calculations (GIAO-HDFT scaling factor for 13C NMR chemical shifts calculation, DOI: 10.1002/poc.1749). We claim to our scaling factor protocol high accuracy and low computational cost. Although behaviour effects were not considered, it also were able achieved great experimental data reproduction.