What procedure should one employ when trying to determine a chemical structure from an NMR spectrum? I have discussed a number of such examples in the past, most recently the procedure by Goodman for dealing with the situation where one has the experimental spectra of 2 diastereomers and you are trying to identify the structures of this pair.1 Now, Goodman provides an extension for the situation where you have a single experimental NMR spectrum and you are trying to determine which of a number of diasteromeric structures best accounts for this spectrum.2 Not only does this prescription provide a means for identifying the best structure, it also provides a confidence level.
The method, called DP4, works as follows. First, perform an MM conformational search of every diastereomer. Select the conformations within 10 kJ of the global minimum and compute the 13C and 1H NMR chemical shifts at B3LYP/6->31G(d,p) – note no reoptimizations! Then compute the Boltzmann weighted average chemical shift. Scale these shifts against the experimental values. You’re now ready to apply the DP4 method. Compute the error in each chemical shift. Determine the probability of this error using the Student’s t test (with mean, standard deviation, and degrees of freedom as found using their database of over 1700 13C and over 1700 1H chemical shifts). Lastly, the DP4 probability is computed as the product of these probabilities divided by the sum of the product of the probabilities over all possible diastereomers. This process is not particularly difficult and Goodman provides a Java applet to perform the DP4 computation for you!
In the paper Smith and Goodman demonstrate that in identifying structures for a broad range of natural products, the DP4 method does an outstanding job at identifying the correct diastereomer, and an even better job of not misidentifying a wrong structure to the spectrum. Performance is markedly better than the typical procedures used, like using the correlation coefficient or mean absolute error. I would strongly encourage those people utilizing computed NMR spectra for identifying chemical structures to considering employing the DP4 method – the computational method is not particularly computer-intensive and the quality of the results is truly impressive.
Afternote: David Bradley has a nice post on this paper, including some comments from Goodman.
References
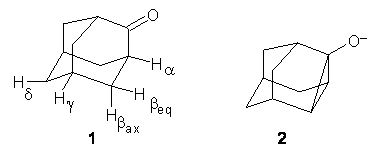
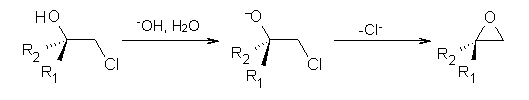
(1) Smith, S. G.; Goodman, J. M., "Assigning the Stereochemistry of Pairs of Diastereoisomers Using GIAO NMR Shift Calculation," J. Org. Chem., 2009, 74, 4597-4607, DOI: 10.1021/jo900408d
(2) Smith, S. G.; Goodman, J. M., "Assigning Stereochemistry to Single Diastereoisomers by GIAO NMR Calculation: The DP4 Probability," J. Am. Chem. Soc., 2010, 132, 12946-12959, DOI: 10.1021/ja105035r