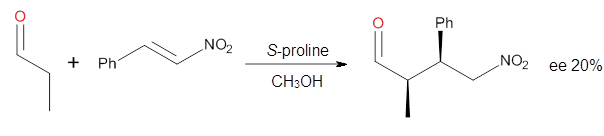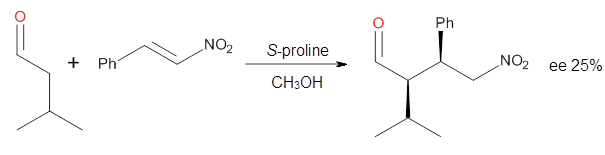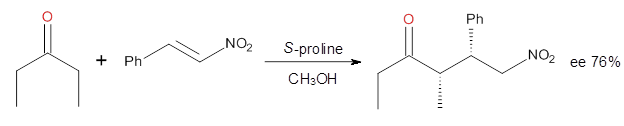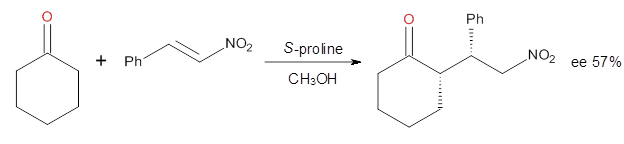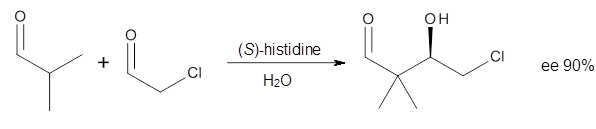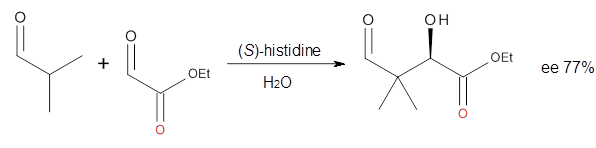Has there been an organic reaction more examined by computational methods than the Diels-Alder reaction? You’d think we would have covered all aspects of this reaction by now, but no, it appears that this reaction remains fertile hunting grounds.
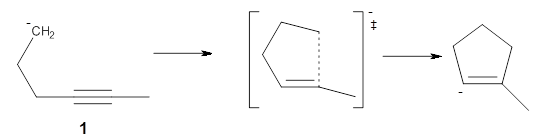
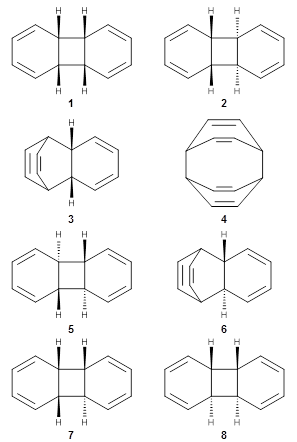
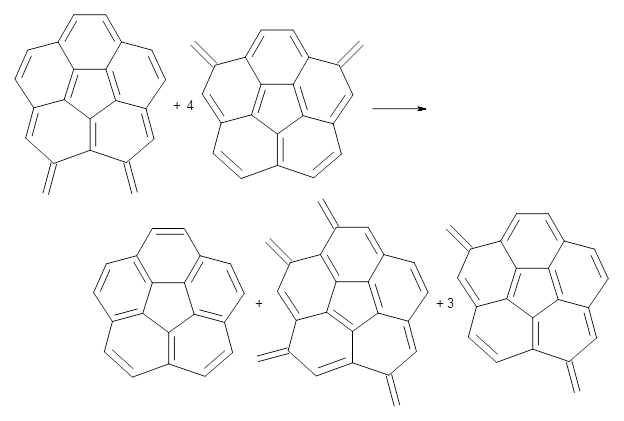
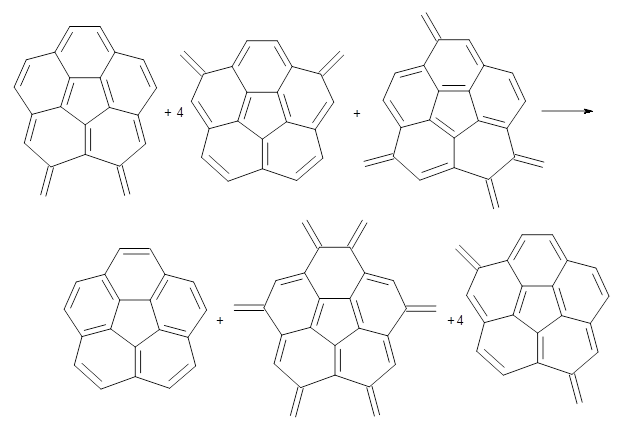
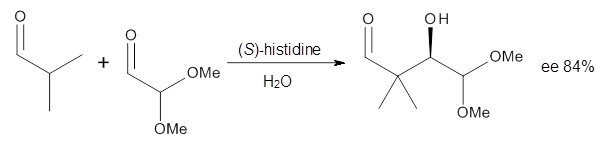
Doubleday and Houk have examined the Diels-Alder reaction with an eye towards its synchronicity,1 an area that Houk has delved into throughout his career. While most experiments show significant stereoselectivity, a few examples display a small amount of stereo loss. Computed transition states tend to have forming C-C bond distances that are similar, though with proper asymmetric substitution, the asymmetry of the TS can be substantial. In this paper,1 they utilize reaction dynamics specifically to assess the time differential between the formation of the two new C-C single bonds. They examined the eight reactions shown below. The first six (R1-R6) have symmetric transition states, though with the random sampling about the TS for the initial condition of the trajectories, a majority of asymmetric starting conditions are used. The last two (R7 and R8) reactions have asymmetric TSs and the random sampling amplifies this asymmetry.
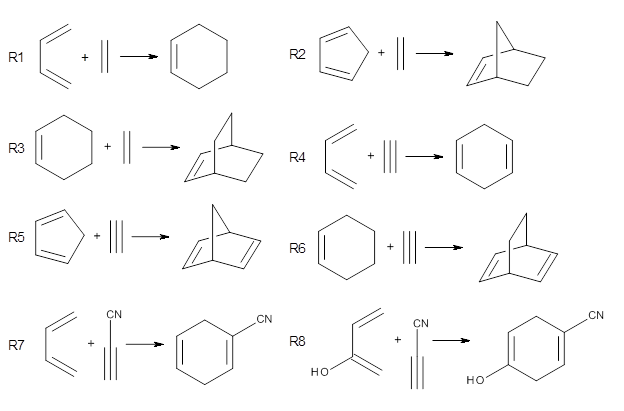
Nonetheless, the results of the dynamics are striking. The time gap, the average time between the formations of the first and second new C-C bond, for R1-R6 is less than 5 fs, much shorter than a C-C vibration. These reactions must be considered as concerted and synchronous. Even the last two reactions (R7 and R8), which are inherently more asymmetric, still have very short time gaps of 15 and 56 fs, respectively. One might therefore reasonably conclude that they too are concerted and synchronous.
There are some exceptions – a few trajectories in the last two reactions involve a long-lived (~1000 fs) diradical intermediate. At very high temperature, about 2% of the trajectories invoke a diradical intermediate. But the overall message is clear: the Diels-Alder reaction is inherently concerted and synchronous.
References
(1) Black, K.; Liu, P.; Xu, L.; Doubleday, C.; Houk, K. N. "Dynamics, transition states, and timing of bond formation in Diels–Alder reactions," Proc. Nat. Acad. Sci. USA, 2012, 109, 12860-12865, DOI: 10.1073/pnas.1209316109