The activation energy for the 5-endo-dig reaction of the anion 1 is anomalously low compared to its 4-endo-dig and 6-endo-dig analogues. Furthermore, the TS is quite early, earlier than might be expected based on the Hammond Postulate. Alabugin and Schleyer have examined this reaction and found some interesting results.1
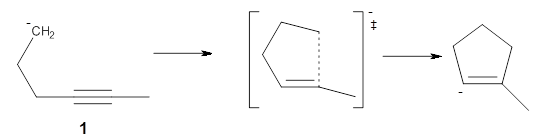
First, NICS(0) values for a series of related intermolecular anionic attack at alkynes show some interesting trends (Table 1). Two of the transition states look like they might be aromatic: the TSs for the 3-exo-dig and the 5-endo-dig reaction have NICS(0) values that are quite negative. However, given the geometry of these TSs, particularly the close proximity of the σ bonds to the ring center, one might be concerned about contamination of these orbitals. So, NICS(0)MOzz computations, which look at the tensor component perpendicular to the ring using just the π-MOs, shows that the 3-exo-dig is likely non-aromatic (NICS(0)MOzz is near zero), the TS for the 4-endo-dig reaction is antiaromatic (NICS(0)MOzz very positive) and the TS for the 5-endo-dig reaction is aromatic (NICS(0)MOzz is very negative. So this last reaction is the first example of an aromatic transition that is not for a pericyclic reaction!
Table 1. NICS(0) and NICS(0)MOzz for the TS of some anionic alkyne cyclizations.
|
|
NICS(0) |
NICS(0)MOzz |
|
|
-19.3 |
-1.6 |
|
|
1.8 |
23.9 |
|
|
-15.2 |
-20.5 |
These authors argue that the reaction of 1 is an “aborted” sigmatropic shift. A normal pericyclic reaction is a single step with a single (concerted) transition state. An interrupted sigmatropic shift has an intermediate that lies higher in energy than the reactants, such as in the Bergman cyclization of an enediyne. The aborted sigmatropic shift has an intermediate that lies lower in energy than the reactants, such as in the cyclization of 1.
References
(1) Gilmore, K.; Manoharan, M.; Wu, J. I. C.; Schleyer, P. v. R.; Alabugin, I. V. "Aromatic Transition States in Nonpericyclic Reactions: Anionic 5-Endo Cyclizations Are Aborted Sigmatropic Shifts," J. Am. Chem. Soc. 2012, 134, 10584–10594, DOI: 10.1021/ja303341b

Henry Rzepa responded on 07 Aug 2012 at 1:43 am #
I love the concept of interrupted and aborted pericyclic reactions, via e.g. biradicals or anions. But I think we should ask questions such as “is this purely a feature of the chosen model?” You may have noticed from my (multiple) comments on Dan Singleton’s anionic reaction of thiolate with an unsaturated ketone that I was questioning whether a model which includes only one of the ions in a pair (in Dan’s case, and also here, the cation is not present) is complete enough to past the test I note above.
It might be that it has been proven that the counter-ion (or solvent) has absolutely no effect on eg the nature of the pericyclic process such as that described above. But I cannot help but reflect that ion-pairs set up (almost by definition) dipole moments. And that dipole moments in small molecules much greater than about 40D may not be physically realistic. And so the system probably reorganises to minimise that dipole (the optimal position of the counter-ion at reactant, transition state and product may differ quite substantially. Or perhaps its location matters not a jot?). Certainly one cannot model this reorganisation with just one ion, and without that one cannot find out if the reorganisation matters or not.
When I first started in this game, in 1974, it was absolutely acceptable to publish an article with the system under study pared down to the minimum. Groups such as Ph, alkyl etc were all replaced by H. And charged species were modelled as that (non zero charges). But what goes into a reaction flask is invariably neutral (apart from some static charge). Should we now make it a mandatory requirement that, like the modern use of the “real substituent” rather than H, we should do as much modelling as is realistic with neutral assemblies? Perhaps we might conclude that classic transition state theory is no longer so useful, and that statistical dynamic approaches are the only way forward? I am not quite ready to abandon TSs yet though.
Oh, one lesson from another molecule, the DNA duplex. One may ask a question such as: is the left or right handed helix most favoured? (answer: it depends on the nature of the base pairs). But what to do about the phosphate anions present? Their negative charges repel, and there is nothing to stop this happening. So add a cation to give an ion-pair! But this gives an unphysical (albeit one with no overall charge) model. One now has to add loads of water to prevent the whole structure from collapsing. Now, as it happens, the structure computed (for four base pairs as a hexa-anion) and the real solvated system with counterions and water are not actually that dissimilar. Perhaps the apparently unphysical model without counterions is actually not that bad, but we had to prove that! And who knows what we might learn from a more complete (neutral) model that the reduced (anionic) model will miss, unless we give it a try.
Igor Alabugin responded on 07 Aug 2012 at 10:33 pm #
Dear Henry,
Thank you for your comment. If I understand it correctly, you are making a general call for the inclusion of counterions in computations of charged species. I am all for it! For reactions in solutions which proceed via contact ions pairs, use of the full model that you advocate would be crucial.
On the other hand, for separated ions pairs where cation and anions are well-solvated and partially shielded, the situation is less obvious and inclusion of explicit solvent molecules may be more helpful, at least in some cases. From yet another perspective, counter-ions in the computational model are obviously not needed for reactions of naked ions in the gas phase. So, I guess the answer would depend on the problem that one is trying to solve and the system that one is trying to model. The focus of our paper was on the fundamental connections between structures of transition states and aromaticity rather than on modeling a specific experimental system. For specific experimental systems, one has to consider ion-pairing, solvation, aggregation. However, even in those cases, the simplest possible model that captures the essence of chemistry and reproduces experimental observations is still the most appealing to me. As it was for Father William of Occam!
Henry Rzepa responded on 14 Aug 2012 at 10:39 am #
The fascinating aspect of pericyclic reactions (real, interrupted or aborted) is there are often multiple ways of looking at them.
Thus one might consider the reaction of 1 to be merely the half way stage of a cycloaddition reaction between ethene and an allenyl anion, continuing on to the cyclised product. So, if 1 were to be more stable than allenyl anion+ethene and also the cyclic vinyl anion product, then it would be an aborted cycloaddition, otherwise it would be an interrupted cycloaddition. A ωb97xd/6-311+g(d,p)/scrf=(cpcm,solvent=water) (OK, I am adding solvent here, which might change things) calculation reveals the following relative free energies: ethene + allenyl anion 0.0, 1 +5.9, the TS for forming the second C-C bond +22.4 and the final cyclic product -26.0 kcal/mol (the TS for the first C-C bond formation to actually form 1 is not yet calculated). So as well as being an aborted sigmatropic shift, the reaction Alabugin and Co. describe could also be regarded as an interrupted (aromatic) cycloaddition reaction! Of course, given my comment about counter-ions above, one does wonder whether their inclusion would change things.
Taking the analogy to the 4-endo-dig reaction (found to be antiaromatic), could this also be regarded as a (interrupted) cycloaddition reaction between an allenyl anion and a carbene? Arrow pushing seems to suggest that this would be a six-electron process, but perhaps two of these electrons are somehow redacted?
Henry Rzepa responded on 14 Aug 2012 at 2:33 pm #
Postscript: If one includes one Na(+) cation in the above reaction, the ethene + sodium allenyl ion pair is 0.0 and the the ion-pair from 1 is +2.5 kcal/mol. So its still an interrupted ion-pair cycloaddition.
Igor Alabugin responded on 15 Aug 2012 at 12:15 am #
Henry,
I agree and I think that your idea of merging several pericyclic reactions together is truly fascinating! It would be very interesting to study it systematically.
I do would like to comment on the following statement though: “So as well as being an aborted sigmatropic shift, the reaction Alabugin and Co. describe could also be regarded as an interrupted (aromatic) cycloaddition reaction! ” This is true but with one important caveat. Let me explain by introducing the four important structures involved in the cyclizations (would be so much easier if I could use a drawing!).
The interrupted cycloaddition would start with ethene and allenyl anion (let’s call them “A”) to give compound 1 as a possible intermediate which, in the absence of the trapping agent, gives the cyclic “5-endo” product (“B”). The latter could rearrange into the product of formal 2,3-Wittig shift (let’s call it “C”) but it does not, because the last step would be thermodynamically favorable (and, hence, is “aborted”).
The interrupted cycloaddition that you suggest corresponds to the A->1->B transformation whereas the aborted sigmatropic shift that we considered corresponds to the 1->B->C transformation. So, the two sequences overlap but do not merge completely. I do agree, of course, that they share the same aromatic TS (1–>B)! What an uncanny coincidence!