Has there been an organic reaction more examined by computational methods than the Diels-Alder reaction? You’d think we would have covered all aspects of this reaction by now, but no, it appears that this reaction remains fertile hunting grounds.
Doubleday and Houk have examined the Diels-Alder reaction with an eye towards its synchronicity,1 an area that Houk has delved into throughout his career. While most experiments show significant stereoselectivity, a few examples display a small amount of stereo loss. Computed transition states tend to have forming C-C bond distances that are similar, though with proper asymmetric substitution, the asymmetry of the TS can be substantial. In this paper,1 they utilize reaction dynamics specifically to assess the time differential between the formation of the two new C-C single bonds. They examined the eight reactions shown below. The first six (R1-R6) have symmetric transition states, though with the random sampling about the TS for the initial condition of the trajectories, a majority of asymmetric starting conditions are used. The last two (R7 and R8) reactions have asymmetric TSs and the random sampling amplifies this asymmetry.
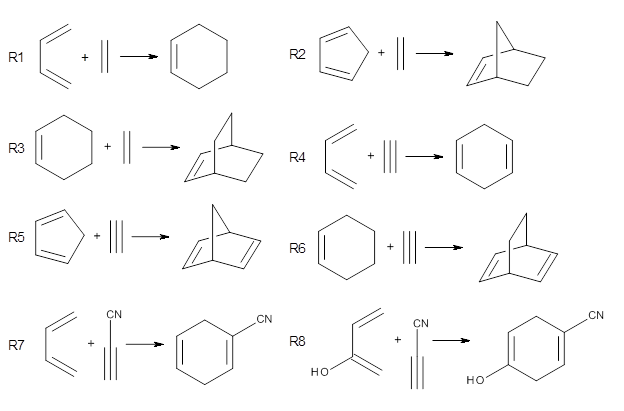
Nonetheless, the results of the dynamics are striking. The time gap, the average time between the formations of the first and second new C-C bond, for R1-R6 is less than 5 fs, much shorter than a C-C vibration. These reactions must be considered as concerted and synchronous. Even the last two reactions (R7 and R8), which are inherently more asymmetric, still have very short time gaps of 15 and 56 fs, respectively. One might therefore reasonably conclude that they too are concerted and synchronous.
There are some exceptions – a few trajectories in the last two reactions involve a long-lived (~1000 fs) diradical intermediate. At very high temperature, about 2% of the trajectories invoke a diradical intermediate. But the overall message is clear: the Diels-Alder reaction is inherently concerted and synchronous.
References
(1) Black, K.; Liu, P.; Xu, L.; Doubleday, C.; Houk, K. N. "Dynamics, transition states, and timing of bond formation in Diels–Alder reactions," Proc. Nat. Acad. Sci. USA, 2012, 109, 12860-12865, DOI: 10.1073/pnas.1209316109

Henry Rzepa responded on 19 Sep 2012 at 3:16 am #
The transition to molecular dynamics is seen appearing everywhere! In my own lectures on pericyclic reactions, I use this example to illustrate asymmetry in bond formation in prosolanapyrone (III) (digital repository link to this calculation can be found here if you want to take a look yourself. I also include a shameless plug for my open-access iPad version of this stuff). But just as the aromaticity of p-cyclophanes is resistant to quite large deformations of the benzene ring, so too I suspect is the concertedness of the themally allowed pericyclic process.
I am also struck that a cheap and cheerful version of molecular dynamics is the intrinsic reaction coordinate, where very often the timings of bond formations in pericyclic processes can also be revealed nicely. Of course, with an IRC one does not get the statistics, just a (probable) single trajectory. It is precisely gathering those statistics which also make dynamics comparatively expensive. I would also comment that just as the structures and reactivity of polybenzenoid aromatics is dominated by the so-called Clar sextet of electrons, so I think this relative synchrony is a feature of six-electron pericyclic processes, such as the Diels-Alder reaction. I strongly suspect that 10 electron thermally allowed pericyclic processes would display nothing like the synchrony that Ken reports for the Diels-Alder.
Of course, thermally forbidden pericyclic processes are quite another game; there intrinsic non-synchrony of bond formations is inherent (and eventually of course losing even the concertedness). Take for example the very simple reaction between ethene and dichlorocarbene (which I believe Ken has studied using dynamics), although here too the time between the two bonds forming may not be long enough to allow molecular vibrations.
So I guess we have another way of expressing the Woodward-Hoffmann rules, namely allowed reactions differ very much less in asynchrony of bond formation (for those that involve more than one bond of course) whereas forbidden reactions demonstrate very high asynchrony. Who knows, perhaps in the future, lecture courses in pericyclic reactions will indeed be taught this way?
Henry Rzepa responded on 19 Sep 2012 at 7:24 am #
As a follow up to the above, I was reminded of a reaction related to Diels-Alder cycloaddition; the cheletropic process where in effect both the bonds form/break to the same atom. The cycloaddition/cheletropic elimination of dichlorocarbene to ethene is a well known reaction, as I noted in the comment above. But what about the six electron equivalent of adding dichlorocarbene to butadiene to form a dichlorocyclopenene. One might intuitively anticipate that it too would form both C-C bonds largely in synchrony. To find out if it does, you will have to see here.