I think most organic chemists hold dear to their hearts the notion that selectivity is due to crossing over different transition states. Readers of my book and this blog know of the many examples where this notion simply is not true (see here). This post discusses yet another example taking place in a seemingly simple reaction.
Singleton has examined the nucleophilic substitution reaction of 1 with sodium tolylsulfide.1 The mono substitution gives potentially two different stereoproducts 2 and 3. The experimental ratio of these products 2:3 is 81:19. (Note that things are a bit more complicated because disubstitution can also occur, but this has been factored into the product ratio.)
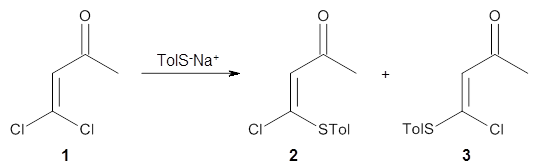
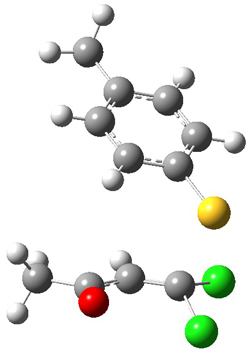
Based on previous literature, this reaction is likely to proceed in a concerted fashion, and so one might anticipate running computations to locate a transition state leading to 2 and a transition state leading to 3. In fact, Singleton finds six different TSs (the lowest energy TS 4 is shown in Figure 1), all within 2 kcal mol-1 of each other at PCM(ethanol)/B3LYP/6-31+G**. However, the intrinsic reaction coordinate going forward from each of these six TSs leads solely to 2; no TS could be located that connects to 3! (Computations were also performed at PCM(ethanol)/M06-2x/6-31+G** which give very similar results.) Classical transition state theory would lead
one to conclude that only 2 should be formed, inconsistent with experiment.
 4 |
 5 |
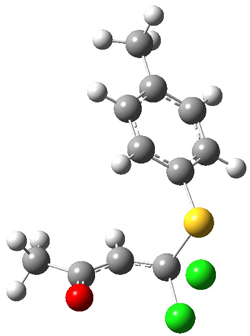
Figure 1. PCM/B3LYP/6-31+G** optimized structures of TSs 4 and 5.
Furthermore, no intermediate could be located. This is consistent with a concerted mechanism. A second transition state was located which interconverts 2 and 3 with the involvement of a chloride – a sort of addition/rotation/elimination process. This TS 5 is also shown in Figure 1.
A direct dynamics study was performed, and 197 trajectories were computed. Of these, 185 trajectories went to product: 156 to 2 and 29 to 3, for a ratio of 84:16 – in amazing agreement with experiment! The product selectivity is due entirely to dynamic effects. In fact, it is one vibrational mode that dictates the product distribution. Essentially, the nature of the rotation about the C=C bond differentiates the eventual route, with a clockwise rotation leading always to 2 and a counterclockwise rotation leading about a third of the time to 3.
References
(1) Bogle, X. S.; Singleton, D. A. "Dynamic Origin of the Stereoselectivity of a Nucleophilic Substitution Reaction," Org. Lett., 2012, 14, 2528-2531, DOI: 10.1021/ol300817a.
InChIs
1: InChI=1S/C4H4Cl2O/c1-3(7)2-4(5)6/h2H,1H3
InChIKey=NXDUHPYJFYSBCT-UHFFFAOYSA-N
2: InChI=1S/C11H11ClOS/c1-8-3-5-10(6-4-8)14-11(12)7-9(2)13/h3-7H,1-2H3/b11-7-
InChIKey=NCXXSKTZGJETLW-XFFZJAGNSA-N
3: InChI=1S/C11H11ClOS/c1-8-3-5-10(6-4-8)14-11(12)7-9(2)13/h3-7H,1-2H3/b11-7+
InChIKey=NCXXSKTZGJETLW-YRNVUSSQSA-N

Henry Rzepa responded on 10 Jul 2012 at 12:21 am #
Stee,
The TS you show indicate that the chlorine has no explicit solvation of its own, i.e. hydrogen bonds from chloride and the ethanol solvent. When we simulated perhaps the most famous solvolysis of all time, Meerwein’s demonstration that isobornyl chloride racemises when placed in o-cresol, and his inference of the intervention of a carbocation, we found we could not get the characteristics correct unless at least two explicit hydrogen bonds were forming to the departing chloride anion (DOI: 10.1021/jo100920e). One has to wonder what effect doing this might have upon the nucleophilic substitution shown here?
Henry Rzepa responded on 11 Jul 2012 at 12:49 am #
Can I point to another example of how solvent may influence the (dynamic) outcome of a reaction. The reaction between a peracid and acetylene sound simple, but in fact is complex. The final products include ketene, but the initial product may be oxirene. The IRC for this reaction looks quite different when computed in a solvent field and the gas phase. In fact the pathway is quite fragile, since it depends also on the substituents on the alkyne. I suspect there may be a few reactions which might be said to be on the edge of chaos, where tiny perturbations have big effects.
We have collected examples of quite a few reactions where either the addition of a continuum solvent field, or alternatively the addition of even a single solvent molecule, can have such a big effect.
Henry Rzepa responded on 11 Jul 2012 at 12:48 pm #
I commented in an earlier post on the importance of a complete model. The simulation above is done as an anionic system; I have to ask: what is the Na+ cationic counter-ion doing whilst the dynamics is controlling the outcome of this reaction? We do not know because its not been included? One might imagine the real system is in fact an ion-pair, and that the characteristics of such an ion-pair (its dipole moment for example) evolve during the dynamical course of the reaction. But if its not included in the model, how are we to know that the computational outcome in its absence is reliable? The Na+ will no doubt attract solvent, and at the start it may be close to the sulfide, and at the end to the chloride. How can that not be a perturbation?
Steven Bachrach responded on 11 Jul 2012 at 1:26 pm #
While I do not want to defend Dan’s work, there are a few things to point out regarding Henry’s comments. First, the solvent is dry ethanol and so there could very well be solvated ions, in which case the sodium gegenion might play almost no role here. Second, while solvent is certainly an important factor, I find it hard to suggest how one might reasonably perform a trajectory study including the solvent. Lastly, one should keep in mind the excellent agreement of the predicted and experimental product distribution here. Is neglect of solvent just fortuitous?
Henry Rzepa responded on 11 Jul 2012 at 2:40 pm #
Steve, you ask to take in mind the excellent agreement of the product distribution. Sadly the computational literature is littered with examples of the right answer for the wrong reason. I am merely suggesting that neglect of both solvent AND counterion seems a significant perturbation from the real system. My experiences with both counter-ion and solvent have suggested that they play a larger role than most people would feel happy neglecting, and that it can often take only a very small perturbation to affect the outcome of a reaction. Thus I have unpublished examples where a change of the continuum solvent field from thf to benzene results in an entirely different product, starting from what looks like a very similar transition state. I note that these examples are also ion-pairs; indeed ion-pairs involving chloride anions.
Henry Rzepa responded on 13 Jul 2012 at 12:35 am #
I would like to add a comment regarding the first line of this post I think most organic chemists hold dear to their hearts the notion that selectivity is due to crossing over different transition states..
The reaction described includes ion-pairs in both reactant (sodium thiolate) and product (sodium chloride), heavily solvated by ethanol. At the transition state, these ion-pairs may well be expressed by a salt-bridge, which involves the formation of unusually strong hydrogen bonds (in comparison to unperturbed H-bonds). Such bridges as a result may easily sustain discrete structures which may resist disruption by the bulk solvent. An example of a salt bridge (actually one with two paths) can be found here, involving as it happens a chloride anion. In the example above, two isomeric salt-bridges are possible, one for each product, and they do differ. One of the bridges for example might have secondary H-bonds to the carbonyl group (structure 2), the other would reasonably be expected to have a different arrangement and hence a (slightly?) different energy. So I think if one includes explicit solvent and counter-ion, it is not unreasonable that the selectivity of this reaction may indeed be due to crossing over different transition states. I am perfectly prepared to accept that dynamics also plays a role in determining selectivity, but I am not yet convinced that the simpler answer (Occam’s razor: different transition states) may not also be an explanation.
We will not know unless such models are investigated. I am trying to track down such structure myself, and will report back if I find any.
Dynamic effects in nucleophilic substitution at trigonal carbon. « Henry Rzepa responded on 15 Jul 2012 at 11:18 pm #
[…] Singleton and co-workers have produced some wonderful work showing how dynamic effects and not just transition states can control the outcome of reactions. Steve Bachrach’s blog contains many examples, including this recent one. […]
Henry Rzepa responded on 15 Jul 2012 at 11:24 pm #
Following up my previous remark, I have collected some structures. My first model was to add two water molecules (representing explicit ethanol) and to balance the charge with a proton (Na+ comes later). This now results in a neutral system (albeit it one with medium sized dipole moments). The bottom line is that interactions to the carbonyl group are not the same for 2 and 3, and if that is true, then there must be two (similar but not identical) transition states. I would be interested to see if the direct dynamics method can be realistically applied to this extended reaction model.
Jim responded on 16 Jul 2012 at 8:50 am #
While there’s no doubt explicit solvation changes the shape of the PES, it’s only helpful if you can actually find TSs leading from 1 to 2 and 1 to 3 that make sense of the selectivity. I thought the point of this is that all the TSs that can be located lead to 2–do your solvated structures change that? If they don’t, then Occam’s razor does not apply.
Henry Rzepa responded on 16 Jul 2012 at 10:50 am #
Replying to Jim; we must always be aware of getting the right result for the wrong reasons. Getting the right result by not including what we know to be present, the counter-ion, or ignoring what must surely be strong hydrogen bonds set up to the solvent, may not be a robust solution. We should ask instead: can we get the right result even if we DO include the counter-ion and solvent but without including dynamics? It may be that this would require a somewhat different model for the entire system. I am exploring with Na(+) at the moment to see if such a straightforward model can be set up.
Or, I am suggesting that before the dynamic (free of counter-ion and explicit solvent) approach is accepted as the only solution, one should also show that the simpler (non-dynamic transition state model which does include counter-ion and explicit solvent) definitely cannot explain the result via differentiation brought about by interaction of the counter-ion/solvent with the carbonyl group.
Henry Rzepa responded on 19 Jul 2012 at 6:58 am #
More progress on the above. I have constructed a new model, formed by adding Na(+) to Bogle and Singleton’s one, a further NaOH, and three water molecules, and embedding this in a continuum solvent field. A transition state, and an IRC following this mode, reveal that the original behaviour (concerted, albeit asynchronous, eviction of chloride rather than formation of an intermediate tetrahedral intermediate) is replicated. However, there seems to be a requirement that the breaking C-Cl bond is favoured by being antiperiplanar to the Ar-S bond. This alignment is in turn enforced by coordination to the carbonyl group. This means that only one of the two C-Cl bonds can be broken using this model. To break the other, it is probably that further NaOH+3(?)H2O needs to be added to the model. Perhaps Occam would indeed be turning in his grave! But thus far, there seems no need for direct dynamics.
Now that coordinates for an extended model with counterion and explicit solvent are available, perhaps Bogle and Singleton nonetheless can try their direct dynamics on it, to see if my hypothesis that antiperiplanar alignment is associated with chloride eviction is indeed true.
Henry Rzepa responded on 14 Aug 2012 at 7:05 am #
An update: The starting hypothesis for my comments above would be that the product distribution in this reaction could be accounted for by two alternative schemes. (a) A single transition state with dynamic trajectories that lead to both products, in the ratio of 5:1, OR (b) two transition states differing in free energy by ~ ΔG 1 kcal/mol, giving the same end result (the classical answer). My comment above strove to ask whether, by adding a counterion and explicit solvent, it might be possible to construct (b), thus providing an alternative explanation to (a). I have now built three further models containing just one stoichiometric Na+ counterion and four explicit hydrogen bonded solvent molecules, and subjected each of these new transition states to IRC analysis. The IRC for all of these lead to just one product, the major one, and none led to the minor product. Whilst this does not preclude the possibility that an as yet undiscovered transition state might in fact reveal an IRC that does lead to the minor product (and one moreover that is only ~1 kcal/mol above the TS leading to the major product) the dynamic model (a) seems to be turning out to be pretty robust thus far!
All the IRC pathways located showed some fascinating geometric phenomena I attribute to stereoelectronic effects, and these effects may turn out to be associated with the dynamic behaviour of this reaction. For example, after the transition state is passed on the IRC, both C-Cl bonds weaken significantly, before one breaks entirely and the other strengthens again. It seems that the stereoelectronic effects are strong enough to over-ride any perturbations caused by the presence of (any non-random distribution of) counterion and solvent. Of course, this conclusion can only be drawn for this specific reaction, and cannot be generalised.
Yet another test of robustness would be to explore the TS models with counterion and solvent (my point above having been that this should be a more complete representation of what is actually in solution) and to show that even for such a model, the computed dynamics for at least one of them still lead to a distribution of two products in the correct observed ratio (unlike the IRCs evaluated for the TS).