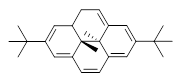
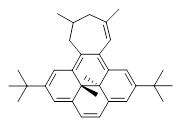
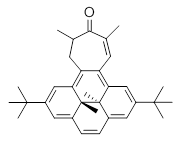
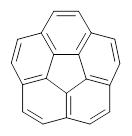
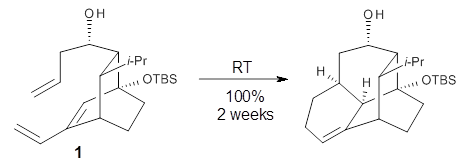
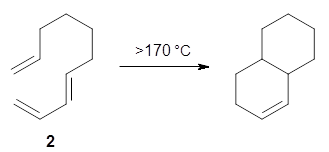
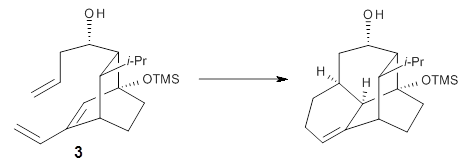
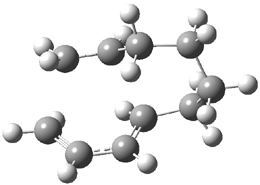
What is the appropriate basis set to use for computing optical rotations? Hedgård, Jensen, and Kongsted examined the optical rotation of 1-6 using B3LYP and CAM-B3LYP at two different wavelengths.1 They examined a series of different basis sets, including the aug-pCS sets2 (developed for NMR computations), the aug-cc-pVXZ series and 6-311++G(3df,3pd). They compared the computed optical rotation with the different basis sets with the value obtained from an extrapolated basis set computation. The mean absolute deviation using either B3LYP or CAM-B3LYP at the two different basis sets are listed in Table 1. The bottom line is that aug-pcS-2 is the preferred method, but this basis set is rather large and computations of big molecules will be difficult. The aug-pcS-1 set is the best choice for large molecules. Errors with the extensive Pople basis set and the aug-cc-pVXZ sets are quite sizable and of concern (especially at the shorter wavelength). It should also be mentioned that even with the largest aug-pcS basis sets extrapolated to the CBS limit, the computed value of the optical rotation of 3 has the wrong sign! Clearly, basis set choice is not the only issue of concern. We remain in need of a robust methodology for computing optical activity.
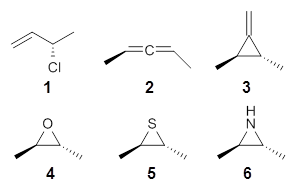
Table 1. Mean absolute deviation of the optical activities of 1-6 evaluated at two wavelengths.
|
|
589.3 nm |
355.0 nm |
||
|
Basis set |
B3LYP |
CAM-B3LYP |
B3LYP |
CAM-B3LYP |
|
aug-pcS-1 |
4.5 |
2.2 |
20.8 |
15.3 |
|
aug-pcS-2 |
1.4 |
1.1 |
4.0 |
1.5 |
|
aug-cc-pVDZ |
15.6 |
13.6 |
62.2 |
144.1 |
|
aug-cc-pVTZ |
3.9 |
6.3 |
9.2 |
37.0 |
|
6-311++G(3df,3pd) |
6.4 |
10.3 |
20.5 |
40.7 |
References
(1) Hedegård, E. D.; Jensen, F.; Kongsted, J. "Basis Set Recommendations for DFT Calculations of Gas-Phase Optical Rotation at Different Wavelengths," J. Chem. Theory Comput. 2012, 8, 4425-4433, DOI: 10.1021/ct300359s
(2) Jensen, F. "Basis Set Convergence of Nuclear Magnetic Shielding Constants Calculated by Density Functional Methods," J. Chem. Theory Comput. 2008, 4, 719-727, DOI: 10.1021/ct800013z