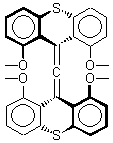
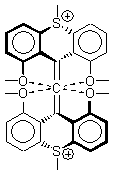
There remains still new territory to explore even with such well-known reactions as the Claisen rearrangement. Jacobsen reports a catalyzed Claisen rearrangement where the catalyst is urea-based.1 Catalyst 1 produces modest to very reasonable % conversion in a series of simple Claisen rearrangements, as shown in Table 1.
|
|
|
|
|
1 |
2 |
|
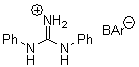
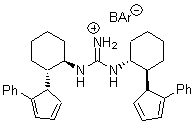
Table 1. Claisen Rearrangements
|
|
% conv |
% conv |
|
|
9 |
15 |
|
|
0 |
72 |
|
|
12 |
76 |
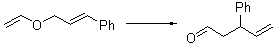
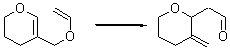
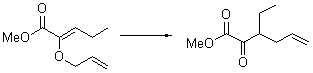
With the chiral catalyst 2, the Claisen rearrangement is both catalyzed and proceeds with large enantiomeric excess, as shown in the representative example Reaction 1.
|
|
Reaction 1 |

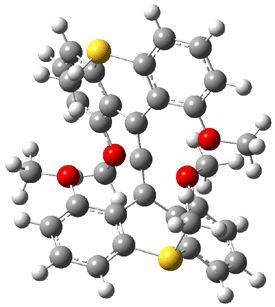
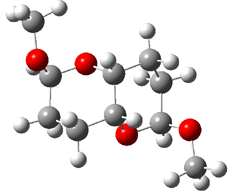
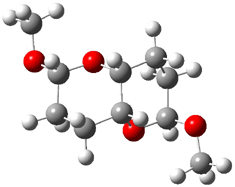
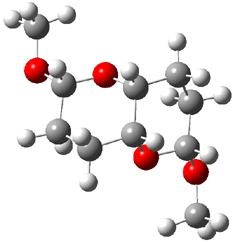
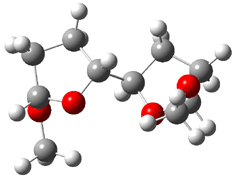
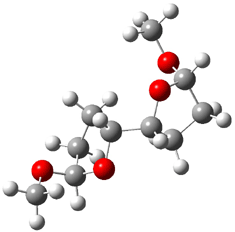
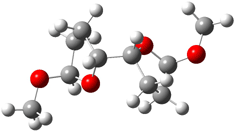
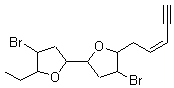
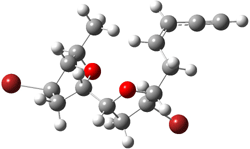
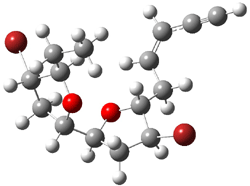
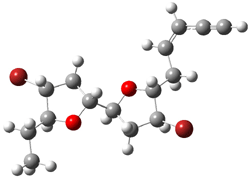
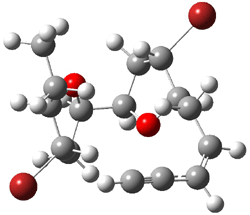
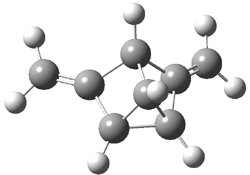
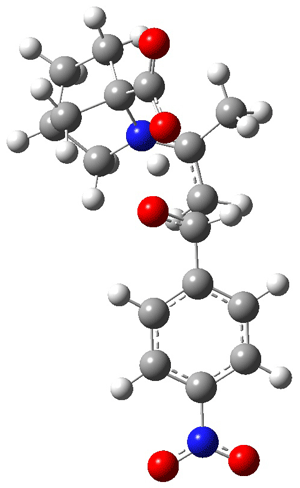
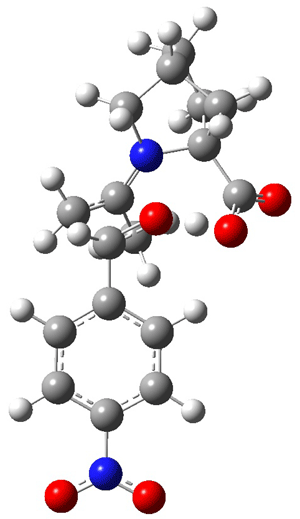
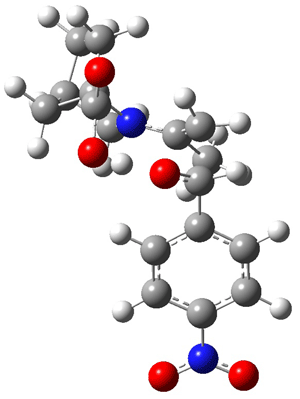
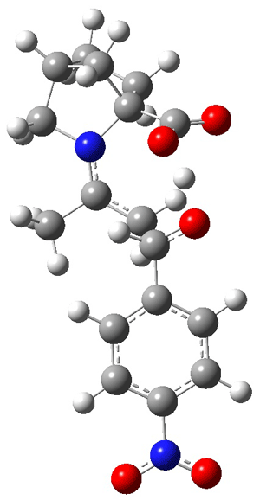
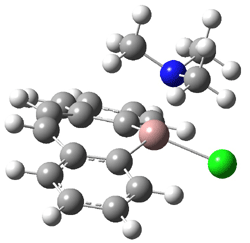
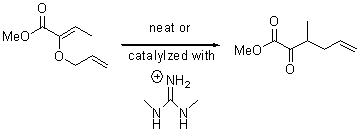
Jacobsen and Uyeda also reported the transition state for a model Claisen rearrangement catalyzed by a model guanidinium ion (Reaction 2) computed at B3LYP/6-31G(d). I have reproduced this calculation and the calculations for the both the catalyzed and uncatalyzed rearrangements, shown in Figure 1. In email communication with Dr. Jacobsen, I was able to confirm these energies against their own unpublished results.
|
|
Reaction 2 |
|
Uncatalyzed Reaction 2 |
|
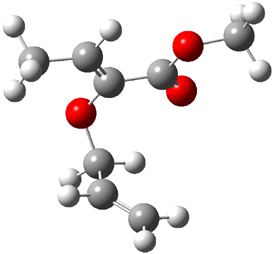 Reactant: 0.0 |
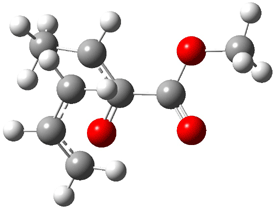 TS: 25.73 |
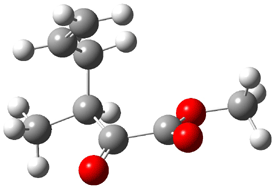 Product 1: -11.04 |
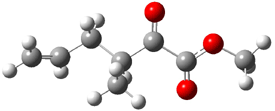 Product 2: -13.69 |
|
Catalyzed Reaction 2 |
|
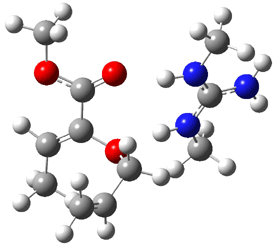 Reactant: 0.0 |
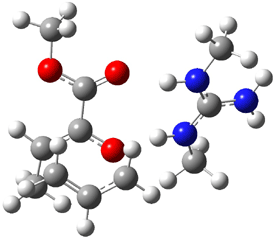 TS: 21.09 |
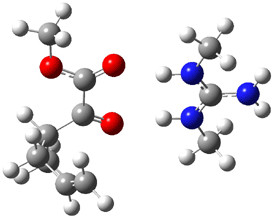 Product 1: -12.36 |
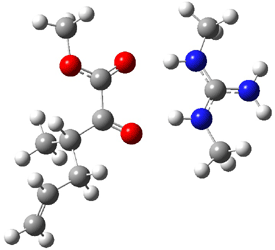 Product 2: -12.80 |
Figure 1. B3LYP/6-31G(d) optimized structures of the critical points of uncatalyzed and catalyzed Reaction 2. Relative energies in kcal mol-1.
The structures show the beneficial hydrogen bonds between the guanidinium anion and the carbonyl oxygens (or the ether oxygen of the reactant). In progressing from the transition state, both reactions first gives Product 1. This product conformer can than rotate to give the lower energy conformer Product 2. The activation energy of the catalyzed reaction is 4.64 kcal mol-1 lower than for the uncatalyzed reaction, demonstrating the benefit of the complexation in the transition state.
I want to thank Dr. Jacobsen and his graduate student Chris Uyeda for sharing their computational results with me for the preparation of this blog post.
References
(1) Uyeda, C.; Jacobsen, E. N., "Enantioselective Claisen Rearrangements with a Hydrogen-Bond Donor Catalyst," J. Am. Chem. Soc., 2008, 130, 9228-9229, DOI: 10.1021/ja803370x.