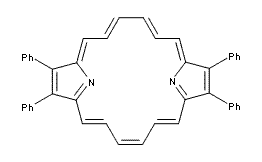
Changing the number of π-electrons in a molecule should alter its aromaticity and antiaromaticity – if the Hückel rule holds! Iron, Cohen and Rybtchinski1 look at an interesting polycyclic aromatic perylene diimide 1 and its dianion and evaluate the aromaticity and reactivity of these species, using both experiment and computation.
|
|
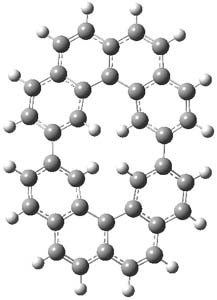
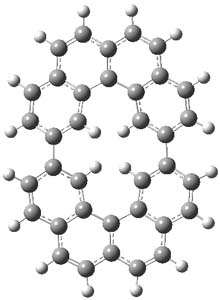
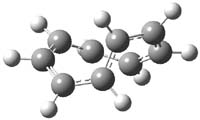
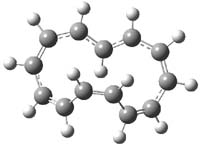
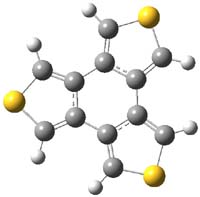
The M06-2x/6-31++G** geometries of 1 and its dianion 12- are shown in Figure 1. 1 can be thought of as two naphthalenyl groups joined together by single bond (their distance is 1.479 Å). In the dianion, the bond distance between the two naphthyl groups (making the central 6-member ring) is shorter, 1.425 Å. In addition the C-C(O) bond of the imide ring shortens from 1.485 to 1.441 Å in going from the neutral to the dianion, with the C=O bond also lengthening, from 1.214 to 1.243 Å. These geometric changes suggest the formation of an aromatic central six-member ring, and perhaps some aromaticity in the diimide rings, too.
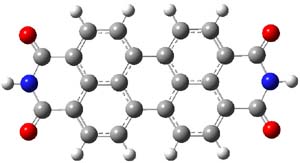 1 |
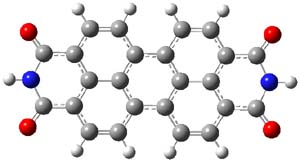 12- |
Figure 1. M06-2x optimized structures of 1 and 12-.
The changes are reflected in the NICS(1) values. In 1, the NICS values for each ring of the naphthyl group is -9.31, as expected for an aromatic 6-member ring. The diimide has a NICS value of -0.01, indicating a non-aromatic ring, and the central ring has a NICS value of +1.78, suggesting some slight antiaromatic character. Significant changes come about with the reduction to the dianion. The central ring now has a NICS value of -9.10, indicating an aromatic ring. The naphthyl rings have NICS values of -3.94 and the diimide ring has a value of -4.53.
So, the dianion 12- species appears to be more aromatic than the neutral 1. This is reflected in its chemistry. The dianion of pegylated 1 is actually stable in water, though it is readily oxidized by air. Its stability in water is understood in terms of the computed reaction energy with water, which is quite positive due to that aromatic stabilization of 12-. This is compared with the computed negative free energies for the reaction of other dianions with water, such as the anthracenyl, and perylenyl dianions.
References
1) Iron, M. A.; Cohen, R.; Rybtchinski, B., "On the Unexpected Stability of the Dianion of Perylene Diimide in Water – A Computational Study," J. Phys. Chem. A, 2011, 115, 2047-2056, DOI: 10.1021/jp1107284
InChIs
1: InChI=1/C24H10N2O4/c27-21-13-5-1-9-10-2-6-15-20-16(24(30)26-23(15)29)8-4-12(18(10)20)11-3-7-14(22(28)25-21)19(13)17(9)11/h1-8H,(H,25,27,28)(H,26,29,30)/f/h25-26H
InChIKey=KJOLVZJFMDVPGB-SPEPDGBUCZ