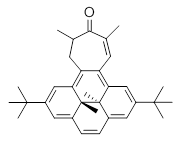
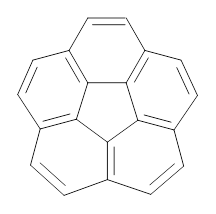
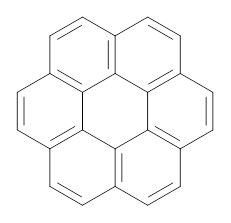
Myśliwiec and Stępień report on a new method for creating buckybowls.1 The usual way had been to build from the inside outward. They opt instead to build from the outside in and have constructed the heterosubstitued bowl chrysaorole 1.
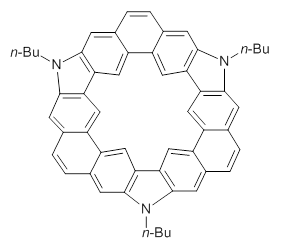
1
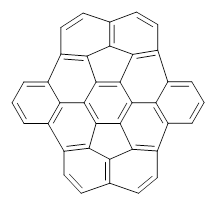
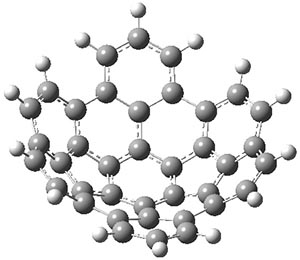
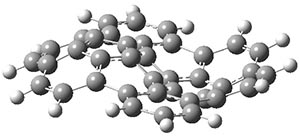
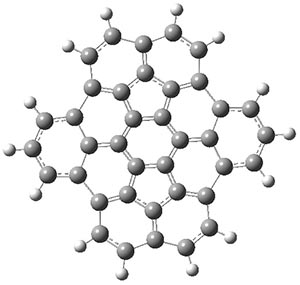
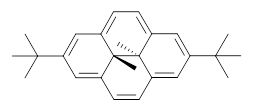
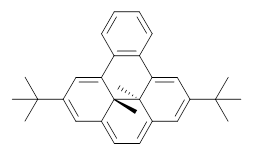
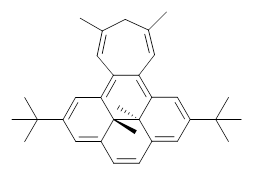
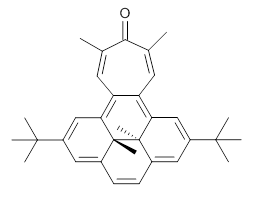
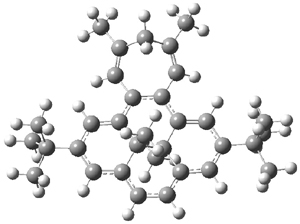
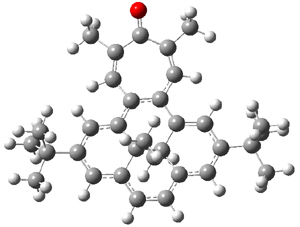
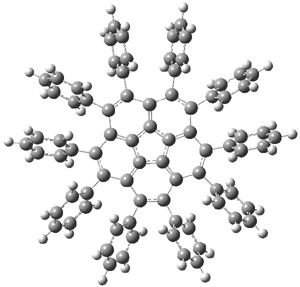
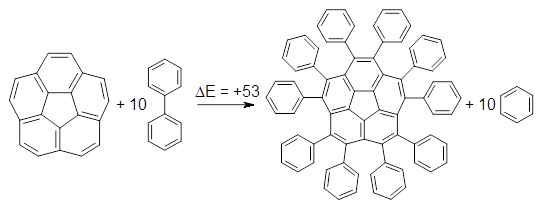
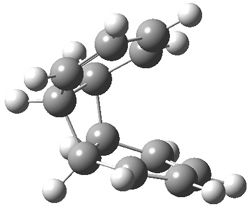
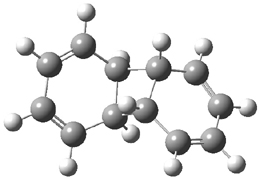
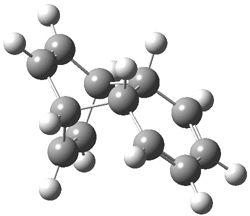
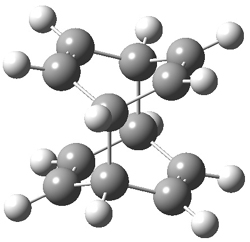
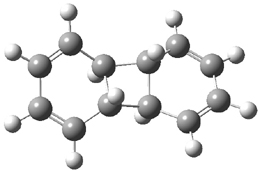
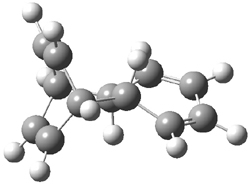
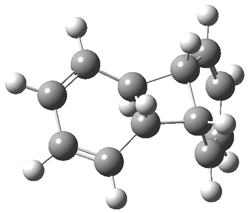
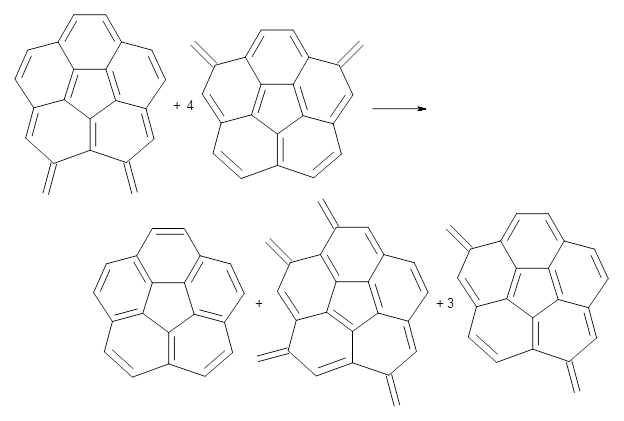
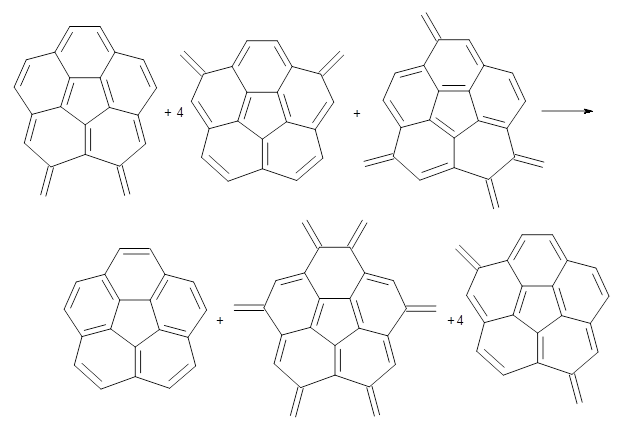
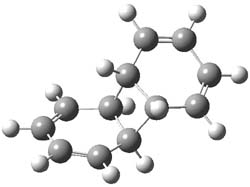
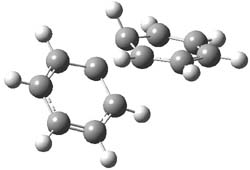
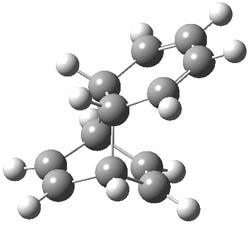
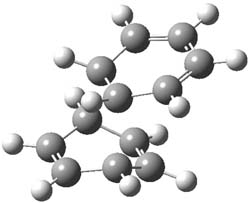
B3LYP/6-31G** optimizations reveal two conformers that are very close in energy: one has the butyl chains outstretched (1a) and one has the butyl arms internal or pendant (1b). These structures are shown in Figure 1. The depth of this bowl (1.96 Å) is quite a bit larger than in corranulene (0.87 Å). The agreement between the computed and experimental 13C and 1H chemical shifts are excellent, supporting the notion that this gas phase geometry is similar to the solution phase structure. Though 1 is strained, 53.4 kcal mol-1 based on B3LYP/6-31G** energies for Reaction 1 (which uses the parent of 1 – replacing the butyl groups with hydrogens), on a per sp2 atom basis, it is no more strained than corranulene.
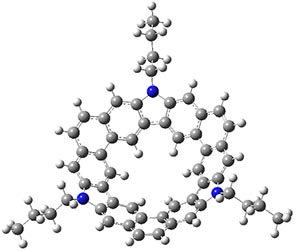 1a |
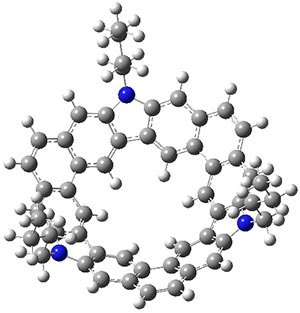 1b |
Figure 1. B3LYP/6-31G** optimized geometries of two conformers of 1.
|
|
Reaction 1 |
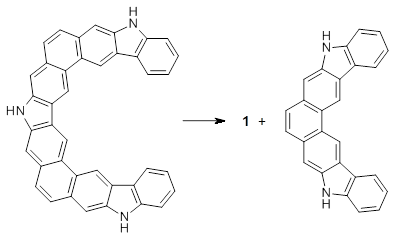
This new synthetic strategy is likely to afford access to more unusual aromatic structures.
References
(1) Myśliwiec, D.; Stępień, M. "The Fold-In Approach to Bowl-Shaped Aromatic Compounds: Synthesis of Chrysaoroles," Angew. Chem. Int. Ed. 2013, 52, 1713-1717, DOI:10.1002/anie.201208547.
InChI
1: InChI=1S/C54H45N3/c1-4-7-16-55-49-19-31-10-12-33-21-51-45-27-39(33)37(31)25-43(49)44-26-38-32(20-50(44)55)11-13-34-22-52-46(28-40(34)38)48-30-42-36(24-54(48)57(52)18-9-6-3)15-14-35-23-53(47(45)29-41(35)42)56(51)17-8-5-2/h10-15,19-30H,4-9,16-18H2,1-3H3
InChIKey=VUUJVETWVYQACL-UHFFFAOYSA-N