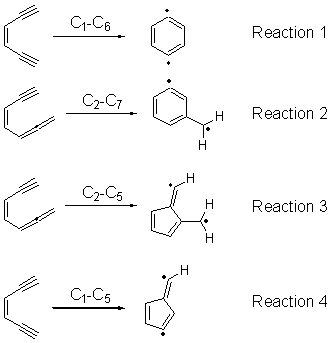
Cyclization of enediynes is thoroughly discussed in Chapter 3.3 of my book. The reaction that started all the excitement is the C1-C6 cyclization (the Bergman cyclization, Reaction 1). Meyers and Saito then proposed the alternative C2-C7 cyclization (Reaction 2), and a variant on this, the Schmittel cyclization (Reaction 3) followed soon thereafter. Now, Pascal completes the theme with a report on the C1-C5 cyclization (Reaction 4).1
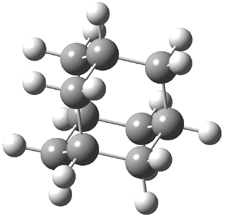
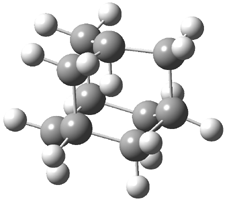
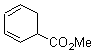
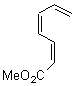
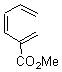
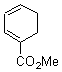
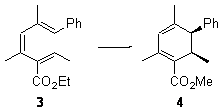
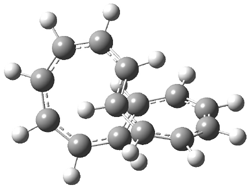
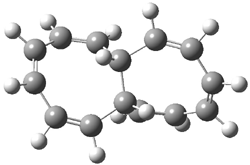
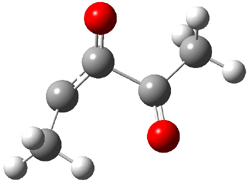
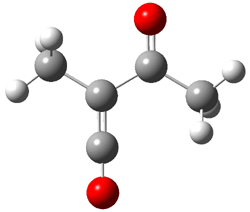
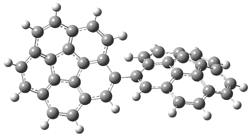
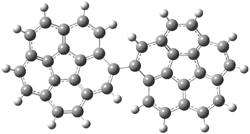
Pascal begins with the assumption that terminal aryl substitution on the enediyne will both (a) inhibit the C1-C6 cyclization due to steric interactions and (b) the C1-C5 cyclization should be enhanced due to stabilization of the radical by the neighboring aryl group. He computed the activation energies of a series of analogues, some of which are listed in Table 1. The transition state structures are shown in Figure 1 for 1b and 1c. Phenyl substitution does accomplish both suggestions: the activation barrier for the Bergman cyclization increases by 4 kcal mol-1, while the barrier for the C1-C5 cyclization is lowered by nearly 6 kcal mol-1. Further substitution of the phenyl ring by either chloro or methyl groups brings the barriers into near degeneracy.
Table 1. RBLYP/6-31G(d) Activation energies (kcal mol-1) for
competing cyclization reactions of substituted enediynes.1
|
|
|||
|
R |
R’ |
Ea(C1-C6) |
Ea(C1-C5) |
|
H (1a) |
H |
24.6 |
37.2 |
|
Phenyl (1b) |
H |
28.7 |
31.4 |
|
2,6-dichlorophenyl (1c) |
H |
30.8 |
31.6 |
|
2,6-dimethylphenyl (1d) |
H |
30.5 |
30.9 |
|
Phenyl (2a) |
Phenyl |
38.5 |
36.3 |
|
2,4,6-trichlorophenyl (2b) |
2,4,6-trichlorophenyl |
43.2 |
38.7 |
|
aComputed at BCCD(T)/cc-pVDZ//-BLYP/6-31G(d). |
|||
 C1-C5 TS of 1b |
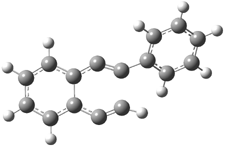 C1-C6 TS of 1b |
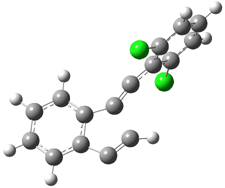 C1-C5 TS of 1c |
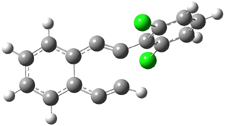 C1-C6 TS of 1c |
Figure 1. RBLYP/6-31G(d) optimized geometries of the C1-C5 and C1-C6 transition states for 1b and 1c.1
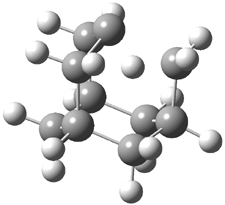
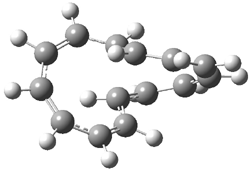
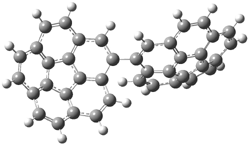
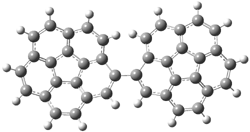
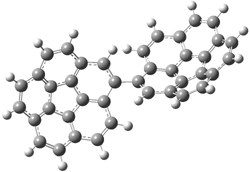
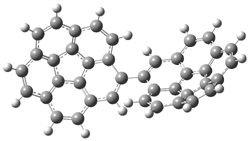
The di-substituted enediynes were examined next. The C1-C5 and C1-C6 transition states for the phenyl (2a) analogue are shown in Figure 2, and the activation energies for it and the 2,4,6-trichlorophenyl (2b) analogue are listed in Table 1. With BLYP, the C1-C5 cyclization is favored by a significant amount over the Bergman cyclization. This may be an overestimation as the BCCD(T)/cc-pVDZ//-BLYP/6-31G(d) computations predict the opposite energy ordering.
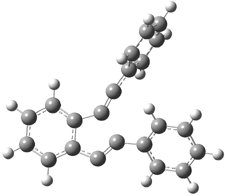 C1-C5 TS of 2a |
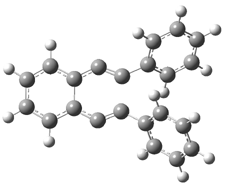 C1-C2 TS of 2a |
Figure 1. RBLYP/6-31G(d) optimized geometries of the C1-C5 and C1-C6 transition states for 2a.1
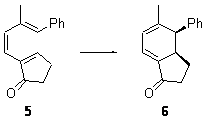
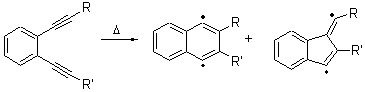
Pascal synthesized 2b and subjected it to thermolysis. Only indenes were obtained, indicative of the C1-C5 cyclization occurring in total preference over the C1-C6 pathway. The presence of 1,4-cyclohexadiene does improve the yields, suggestive that the transfer hydrogenation mechanism may be operative. However, when the reaction is done in the absence of 1,4-cyclohexadiene and at lower temperature (180 °C), the C1-C5 cyclization is still observed and no Bergman cyclization is seen. It appears that C1-C5 cyclization of enediynes is a viable reaction.
References
(1) Vavilala, C.; Byrne, N.; Kraml, C. M.; Ho, D. M.; Pascal, R. A., "Thermal C1-C5 Diradical Cyclization of Enediynes," J. Am. Chem. Soc. 2008, 130, 13549-13551, DOI: 10.1021/ja803413f.
InChIs
1a: InChI=1/C10H6/c1-3-9-7-5-6-8-10(9)4-2/h1-2,5-8H
InChIKey=CBYDUPRWILCUIC-UHFFFAOYAY
1b: InChI=1/C16H10/c1-2-15-10-6-7-11-16(15)13-12-14-8-4-3-5-9-14/h1,3-11H
InChIKey=FFEGFMOHMPSHTK-UHFFFAOYAQ
1c: InChI=1/C16H8Cl2/c1-2-12-6-3-4-7-13(12)10-11-14-15(17)8-5-9-16(14)18/h1,3-9H
InChIKey=ZQRAACNBGPDESE-UHFFFAOYAV
1d: InChI=1/C18H14/c1-4-16-10-5-6-11-17(16)12-13-18-14(2)8-7-9-15(18)3/h1,5-11H,2-3H3
InChIKey=XGUCEMJKUJLOHZ-UHFFFAOYAZ
2a: InChI=1/C22H14/c1-3-9-19(10-4-1)15-17-21-13-7-8-14-22(21)18-16-20-11-5-2-6-12-20/h1-14H
InChIKey=XOJSMLDMLXWRMT-UHFFFAOYAD
2b: InChI=1/C22H8Cl6/c23-15-9-19(25)17(20(26)10-15)7-5-13-3-1-2-4-14(13)6-8-18-21(27)11-16(24)12-22(18)28/h1-4,9-12H
InChIKey=FNGRRGHMCFPDDG-UHFFFAOYAU