A computational study of addition of singlet carbenes to bicyclobutanes reveals another potential energy surface where dynamics may be active. Rablen, Jones and co-workers examined the reaction of dichlorocarbene with bicyclobutane 1 and 1,2,2-trimethylbicyclobutane 2 (Reactions 1 and 2) using a number of computational techniques.1
|
|
Reaction 1 |
|
|
Reaction 2 |


For reaction 1, they identified three reaction pathways. The first two involve the carbene approaching along the central C-C bond. Path A (Scheme 1) involves a single transition state that leads to product 3, with a barrier of 8.4 kcal mol-1. The second pat (pathway B), leads to critical point 4, which is a transition state at HF/6-31G* and QCISD/6-31G* but is a local minimum at CCSD/6-31G*. This minimum however is very shallow, and vibrational energy will exceed the barriers about it. Both pathways indicate an asynchronous but concerted reaction. The last pathway (C) is for insertion of the carbine into the bridgehead C-C bond, leading to the bicyclo product 5. This barrier is very high, 27 kcal mol-1, and so this path is unlikely to be competitive.
|
|
Path A |
|
|
Path B |
|
|
Path c |



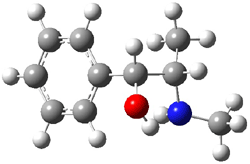
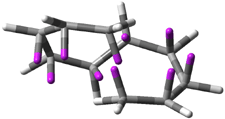
Experimental study of Reaction 2 showed that only 6 is produced.2 Rablen and Jones identified six pathways where the carbene attacks 2 along the bridgehead bond (analogous to Paths A and B, except there are three rotamers and the attack can be at either bridgehead carbon) and the insertion path that leads to 8. Once again, this last pathway has a very large barrier and is non-competitive. Attack at the unsubstituted bridgehead carbons is favored over attack at the methyl-substituted bridgehead by 2-3 kcal mol-1. The path that leads directly to 7 has a slightly lower barrier (0.4 kcal mol-1) than the path that leads directly to 8. The analog of Path B leads here to a true intermediate 9 through a barrier 0.4 kcal mol-1 higher than the barrier that leads to 7. This intermediate is shown in Figure 1.
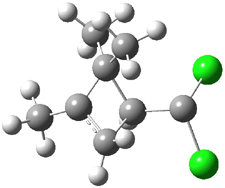 |
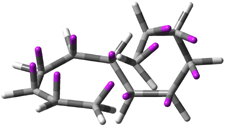
Figure 1. CCSD/6-31G* structure of intermediate 9.1
The energies of the barriers suggest that 7 will be the major product, but not the exclusive product. Rablen and Jones point out that intermediate 9 lies in a very shallow plateau and exit from this intermediate can lead to either 7 or 8. This sort of potential energy surface has been implicated in reactions that exhibit non-statistical behavior indicative of dynamic effects (see Chapter 7 of my book). Rablen and Jones speculate that dynamics might be dictating the product distribution in Reaction 2 as well. Confirmation awaits a molecular dynamics study.
References
(1) Rablen, P. R.; Paiz, A. A.; Thuronyi, B. W.; Jones, M., "Computational Investigation of the Mechanism of Addition of Singlet Carbenes to Bicyclobutanes," J. Org. Chem. 2009, DOI: 10.1021/jo900485z
(2) Jackson, J. E.; Mock, G. B.; Tetef, M. L.; Zheng, G.-x.; Jones, M., "Reactions of carbenes with bicyclobutanes and quadricyclane : Cycloadditions with two σ bonds," Tetrahedron 1985, 41, 1453-1464, DOI: 10.1016/S0040-4020(01)96386-0.
InChIs
1: InChI=1/C4H6/c1-3-2-4(1)3/h3-4H,1-2H2
InChIKey=LASLVGACQUUOEB-UHFFFAOYAV
2: InChI=1/C7H12/c1-6(2)5-4-7(5,6)3/h5H,4H2,1-3H3
InChIKey=GJMVYBBYZUWWLJ-UHFFFAOYAI
3: InChI=1/C5H6Cl2/c1-2-3-4-5(6)7/h2,4H,1,3H2
InChIKey=FGUOQAVVVDPABB-UHFFFAOYAR
5: InChI=1/C5H6Cl2/c6-5(7)3-1-4(5)2-3/h3-4H,1-2H2
InChIKey=SUZACPSWEYRCBD-UHFFFAOYAW
6: InChI=1/C8H12Cl2/c1-6(2)8(3,4)5-7(9)10/h5H,1H2,2-4H3
InChIKey=QIFFCMZJZYIIBA-UHFFFAOYAZ
7: InChI=1/C8H12Cl2/c1-6(2)7(3)4-5-8(9)10/h5H,4H2,1-3H3
InChIKey=MOELQSRRCNAPQV-UHFFFAOYAX
8: InChI=1/C8H12Cl2/c1-6(2)5-4-7(6,3)8(5,9)10/h5H,4H2,1-3H3
InChIKey=PRCOWYZGJRWGOB-UHFFFAOYAP