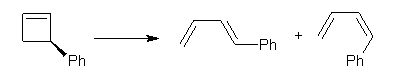
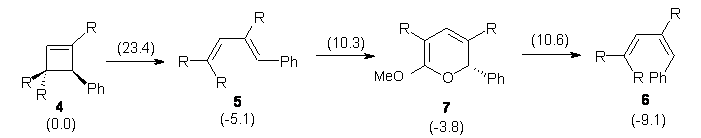
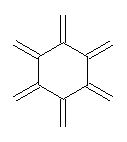
Conjugated alkenes have played a major role in conceptualizing organic chemistry. Linear and cyclic unbranched conjugated alkenes have been well studied; the latter class comprising the aromatic and antiaromatic annulenes. The cyclic branched conjugate alkenes are known as radialenes and have been subject of some study. But the last category, the linear branched conjugated alkenes have been overlooked. Paddon-Row and Sherburn1 now report a general synthetic method for preparing these species, which they call dendralenes, see Scheme 1.
Scheme 1. Classes of conjugated alkenes
|
|
|
|
|
|
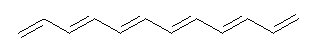
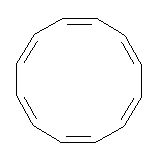
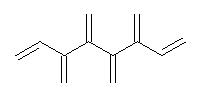
The dendralenes fall into two groups – those with an odd number of double bonds and those with an even number. While the UV/Vis absorption maximum redshifts with increasing length, the molar extinctions coefficients are relatively constant for the odd denralenes but it increases by about 10,000 within the even dendralene family. The Diels-Alder chemistry is even more distinctive: the odd dendralenes react rapidly with an electron deficient dienophile (N-methylmaleimide), with rates decreasing slightly with increasing size, but the even dendralenes are significantly more sluggish.
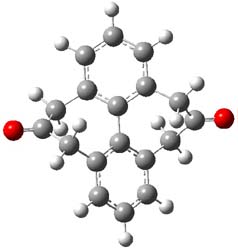
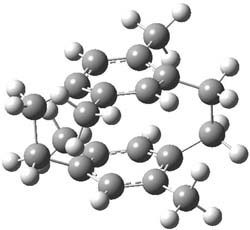
The optimized B3LYP/6-31G(d) geometries of the lowest energy conformers of the [3]- to [8]dendralenes are shown in Figure 1. There are three types of butadiene fragments present in these structures: (a) near planar s-trans arrangement, (b) near perpendicular arrangement of the two double bonds, and (c) s-cis arrangement with the dihedral angle about 40°. The even dendralenes have only the first two type: alternating planar butadiene fragment that are more-or-less orthogonal to each other. The odd dendralenes all have at least one s-cis arrangement. Paddon-Row and Sherburn suggest that since the s-cis arrangement is necessary for the diene component of the Diels-SAlder reaction, the odd dendralenes are more reactive than the even ones since they have this arranegement in their ground state conformations, while the even dendralenes will have to react out of a higher energy conformation. This is a nice explanation readily formulated from simple computations.
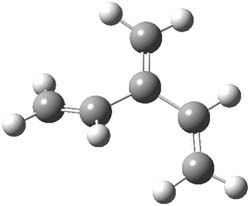 [3]dendralene |
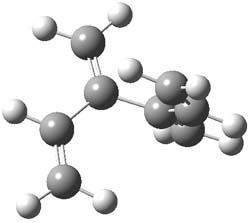 [4]dendralene |
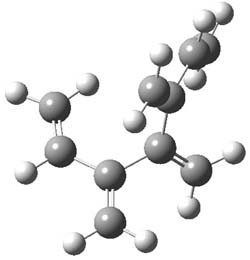 [5]dendralene |
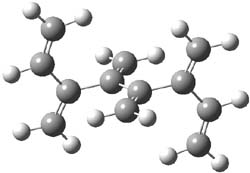 [6]dendralene |
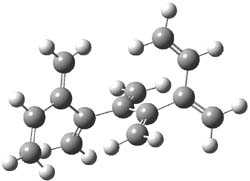 [7]dendralene |
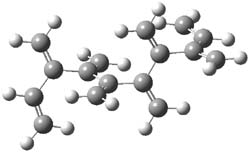 [8]dendralene |
Figure 1. B3LYP/6-31G(d) optimized structures of [3]- to [8]dendralene.1
References
(1) Payne, A. D.; Bojase, G.; Paddon-Row, M. N.; Sherburn, M. S., "Practical Synthesis of the Dendralene Family Reveals Alternation in Behavior," Angew. Chem. Int. Ed. 2009, 48, 4836-4839, DOI: 10.1002/anie.200901733
InChIs
[3]dendralene: InChI=1/C6H8/c1-4-6(3)5-2/h4-5H,1-3H2
InChIKey=VXBVLYQDVVHAHZ-UHFFFAOYAS
[4]dendralene: InChI=1/C8H10/c1-5-7(3)8(4)6-2/h5-6H,1-4H2
InChIKey=DMCINEDFOKMBFI-UHFFFAOYAV
[5]dendralene: InChI=1/C10H12/c1-6-8(3)10(5)9(4)7-2/h6-7H,1-5H2
InChIKey=XEZCEXNNZGLEHB-UHFFFAOYAM
[6]dendralene: InChI=1/C12H14/c1-7-9(3)11(5)12(6)10(4)8-2/h7-8H,1-6H2
InChIKey=RBABOPLFRQKABD-UHFFFAOYAA
[7]dendralene: InChI=1/C14H16/c1-8-10(3)12(5)14(7)13(6)11(4)9-2/h8-9H,1-7H2
InChIKey=ZIBYAXDRKFGSBF-UHFFFAOYAH
[8]dendralene: InChI=1/C16H18/c1-9-11(3)13(5)15(7)16(8)14(6)12(4)10-2/h9-10H,1-8H2
InChIKey=YWPORNAHEZCVCQ-UHFFFAOYAR