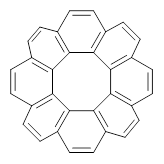
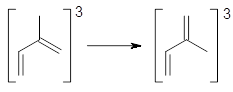
The role of dispersion in large systems is increasingly recognized as critical towards understanding molecular geometry. An interesting example is this study of acene dimers by Grimme.1 The heptacene and nonacene dimers (1 and 2) were investigated with an eye towards the separation between the “butterfly wings” – is there a “stacked” conformation where the wings are close together, along with the “open” conformer?
|
|
|
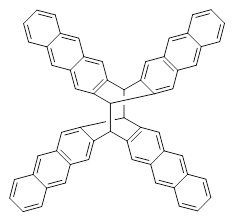
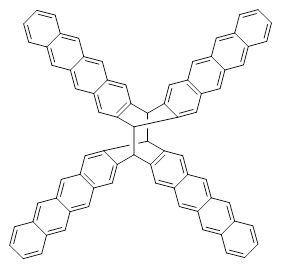
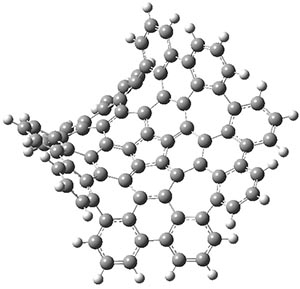
The LPNO-CEPA/CBS potential energy surface of 1 shows only a single local energy minima, corresponding to the open conformer. B3LYP-D3 and B3LYP-NL, two different variations of dealing with dispersion (see this post), do a reasonable job at mimicking the LPNO-CEPA results, while MP2 indicates the stacked conformer is lower in energy than the open conformer.
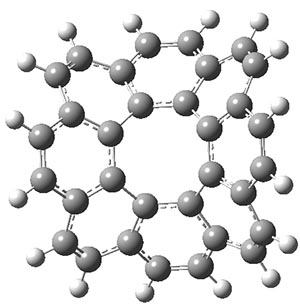
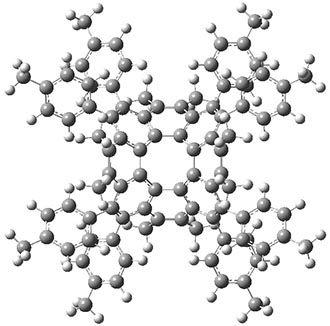
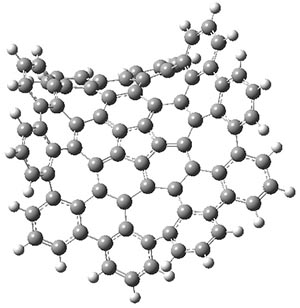
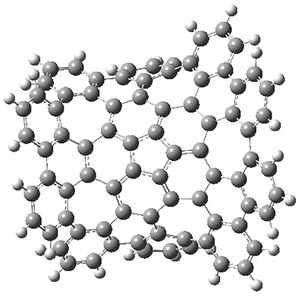
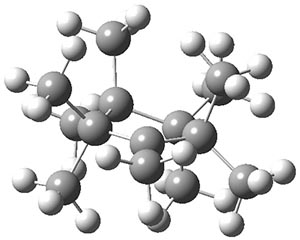
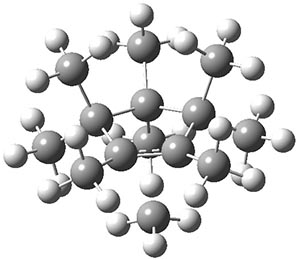
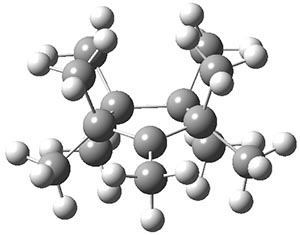
B3LYP-D3 predicts both conformers for the nonacene dimer 2, and the optimized structures are shown in Figure 2. The stacked conformer is slightly lower in energy than the open one, with a barrier of about 3.5 kcal mol-1. However in benzene solution, the open conformer is expected to dominate due to favorable solvation with both the interior and exterior sides of the wings.
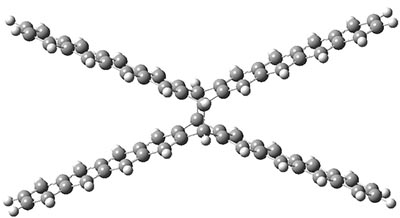 open |
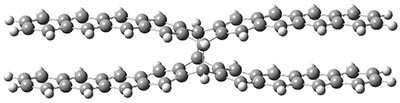 stacked |
Figure 1. B3LYP-D3/ef2-TZVP optimized structures of the open and stacked conformations of 2.
References
(1) Ehrlich, S.; Bettinger, H. F.; Grimme, S. "Dispersion-Driven Conformational Isomerism in σ-Bonded Dimers of Larger Acenes," Angew. Chem. Int. Ed. 2013, 41, 10892–10895, DOI: 10.1002/anie.201304674.
InChIs
1: InChI=1S/C60H36/c1-2-10-34-18-42-26-50-49(25-41(42)17-33(34)9-1)57-51-27-43-19-35-11-3-4-12-36(35)20-44(43)28-52(51)58(50)60-55-31-47-23-39-15-7-5-13-37(39)21-45(47)29-53(55)59(57)54-30-46-22-38-14-6-8-16-40(38)24-48(46)32-56(54)60/h1-32,57-60H
InChIKey=OYVUURMCCBPDLI-UHFFFAOYSA-N
2: InChI=1S/C76H44/c1-2-10-42-18-50-26-58-34-66-65(33-57(58)25-49(50)17-41(42)9-1)73-67-35-59-27-51-19-43-11-3-4-12-44(43)20-52(51)28-60(59)36-68(67)74(66)76-71-39-63-31-55-23-47-15-7-5-13-45(47)21-53(55)29-61(63)37-69(71)75(73)70-38-62-30-54-22-46-14-6-8-16-48(46)24-56(54)32-64(62)40-72(70)76/h1-40,73-76H
InChIKey=FTSMWBVFVPAXOB-UHFFFAOYSA-N