The nine methyl groups of nonamethylcyclopentyl cation 1 all interconvert with a barrier of 7 kcal mol-1. However, at low temperature only partial scrambling occurs: there are two sets of methyl groups, one containing five groups and the other containing four methyl groups. The barrier for this scrambling is only 2.5 kcal mol-1. While this behavior was found more than 20 years ago, Tantillo and Schleyer1 only now have offered a complete explanation.
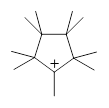
1
The ground state structure of 1 is shown in Figure 1 and has C1 symmetry. The two pseudo-axial methyl groups adjacent to the cationic center show evidence of hyperconjugation: long C-C bonds and Me-C-C+ angles of 100°.
The transition state TS1¸also in Figure 1, is of Cs symmetry. This transition state leads to interchange of the pseudo-axial methyls, and interchange of the pseudo-equatorial methyls, but no exchange between the members of these two groups. The M06-2x/6-31+G(d,p) and mPW1PW91/6-31+G(d,p) estimate of this barrier is 1.5 and 2.5 kcal mol-1, respectively. This agrees well with the experiment.
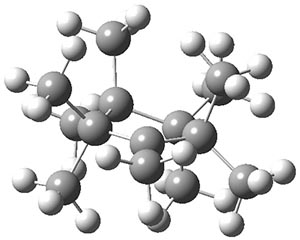 1 |
|
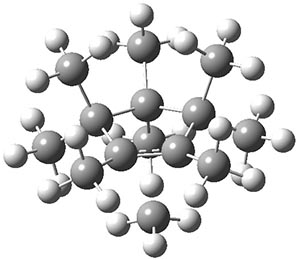 TS1 |
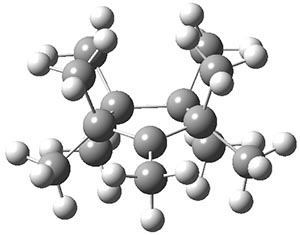 TS2 |
Figure 1. B3LYP/6-3+G(d,p) optimized geometries.
A second transition state TS2 was found and it corresponds with a twisting motion that interconverts an axial methyl with an equatorial methyl. This TS has Cs symmetry (shown in Figure 1) and the eclipsing interaction give rise to a larger barrier: 7.3 (M06-2x/6-31+G(d,p)) and 6.7 kcal mol-1 (mPW1PW91/6-31+G(d,p)). So twisting through TS2 and scrambling through TS1 allows for complete exchange of all 9 methyl groups.
An interesting point also made by these authors is that these three structures represent the continuum of cationic structure: a classical (localized) cation in TS2, a bridged structure in TS1 and hyperconjugated cation in 1.
References
(1) Tantillo, D. J.; Schleyer, P. v. R. “Nonamethylcyclopentyl Cation Rearrangement Mysteries Solved,” Org. Lett. 2013, 15, 1725-1727, DOI: 10.1021/ol4005189.
InChIs
1: InChI=1S/C14H27/c1-10-11(2,3)13(6,7)14(8,9)12(10,4)5/h1-9H3/q+1
InChIKey=WUGVCUSQGLXERW-UHFFFAOYSA-N

Alexander Genaev responded on 29 Jul 2013 at 12:48 am #
There is another explanation for the experimental data. Complete exchange of all 9 methyl groups may occur via scrambling through boatlike TS1 (migration of the pseudo-axial methyls) and then via scrambling through chairlike TS3 (migration of the pseudo-equatorial methyls). On DFT level TS3 is higher than TS2, but on MP2 level vice versa.
Alexander Genaev responded on 29 Jul 2013 at 1:39 am #
Relative to 1
DFT/PBE/L1 energy: 3.1 (TS1), 6.4 (TS2), 8.3 (TS3) kcal/mol
riMP2/L1 energy: 0.3 (TS1), 8.6 (TS2), 5.7 (TS3) kcal/mol
Henry Rzepa responded on 30 Jul 2013 at 11:29 pm #
Are there any IRC profiles for TS1-3? I guess that IRC at MP2 is not feasible, even with ri?
Henry Rzepa responded on 31 Jul 2013 at 12:14 am #
Some more observations.
1. The geometries are computed at the B3LYP level. This is a very crowded molecule, a type that B3LYP (with, inter alia, its lack of dispersion) happens not to handle very well.
2. The activation enthalpies (+ZPE) are computed for the gas phase, whereas the NMR measurements relate to free energies of activation for SbF5 ion pairs in a solvent of high dielectric.
There will certainly be a lot of cancellation of error, but it would be nice to see if a dispersion and continuum-solvent corrected intrinsic reaction coordinate profile (IRC) deriving from the ion-pair geometries for TS1, TS2 and TS3 (as added by Alex) at e.g a triple-ζ level sustains these fascinating interpretations.