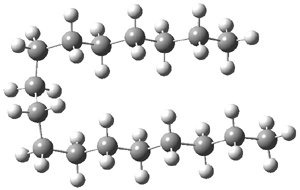
I have written a number of posts discussing long C-C bonds (here and here). What about very long bonds between carbon and a heteroatom? Well, Mascal and co-workers1 have computed the structures of some oxonium cations that express some very long C-O bonds. The champion, computed at MP2/6-31+G**, is the oxatriquinane 1, whose C-O bond is predicted to be 1.602 Å! (It is rather disappointing that the optimized structures are not included in the supporting materials!) The long bond is attributed not to dispersion forces, as in the very long C-C bonds (see the other posts), but rather to σ(C-H) or σ(C-C) donation into the σ*(C-O) orbital.
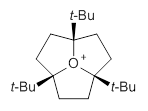
1
Inspired by these computations, they went ahead and synthesized 1 and some related species. They were able to get crystals of 1 as a (CHB11Cl11)– salt. The experimental C-O bond lengths for the x-ray crystal study are 1.591, 1.593, and 1.622 Å, confirming the computational prediction of long C-O bonds.
As an aside, they also noted many examples of very long C-O distances within the Cambridge
Structural database that are erroneous – a cautionary note to anyone making use of this database to identify unusual structures.
References
(1) Gunbas, G.; Hafezi, N.; Sheppard, W. L.; Olmstead, M. M.; Stoyanova, I. V.; Tham, F. S.; Meyer, M. P.; Mascal, M. "Extreme oxatriquinanes and a record C–O bond length," Nat. Chem. 2012, 4, 1018-1023, DOI: 10.1038/nchem.1502
InChIs
1: InChI=1S/C21H39O/c1-16(2,3)19-10-12-20(17(4,5)6)14-15-21(13-11-19,22(19)20)18(7,8)9/h10-15H2,1-9H3/q+1/t19-,20+,21-
InChIKey=VTBHIDVLNISMTR-WKCHPHFGSA-N