The structure of [2,2]paracyclophane 1 has been somewhat controversial for some time. Early x-ray structures indicated that the molecule was quite symmetric, D2h with the phenyl rings and the ethyl bridges eclipsed. Subsequent low-T experiments suggested a lower symmetry form D2 with a twist that relieves some of the unfavorable eclipsing interactions in the ethano bridges. High-level computations by Grimme1 and then some by myself2 indicated that the D2 structure is the lowest energy conformation, with however a low barrier through the D2h structure.
The suggestion of the D2 minimum was vehemently criticized by Dodziuk, et al. on the basis of NMR analysis.3
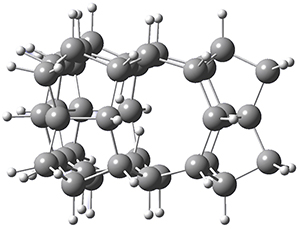
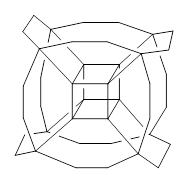
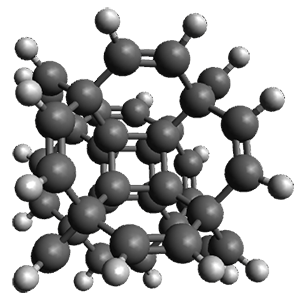
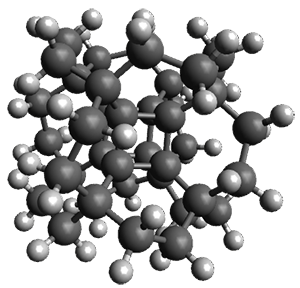
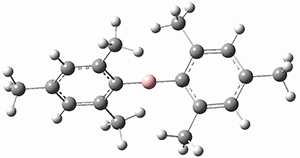
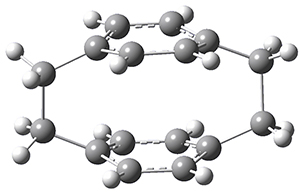
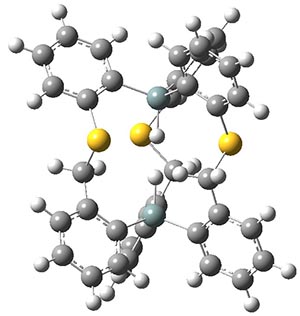
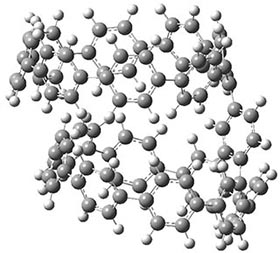
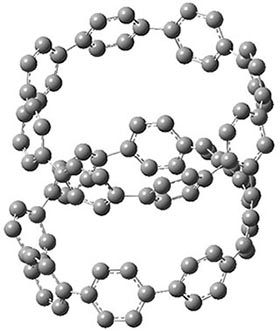
Now, a low temperature x-ray experiment of 1 brings clarity to the situation.4 (The introduction provides a nice summary of the previous 70 year history regarding the structure of 1.) At temperatures below 45 K, 1 is found as a single structure of D2 symmetry (with space group P4n2). The structure is shown in Figure 1. A phase change occurs at about 45 K, and above 60 K the crystal has P42/mnm symmetry. The structure of 1 at the high temperature appears as D2h with somewhat broader thermal motion of the ethano carbons than the phenyl carbons. The low T structure is in excellent accord with the previous theoretical studies, and the phase transition helps bring into accord all of the previous x-ray crystallographic work.
Figure 1. X-ray structure at 15K of 1.
References
(1) Grimme, S. "On the Importance of Electron Correlation Effects for the π-π Interactions in Cyclophanes," Chemistry Eur. J. 2004, 10, 3423-3429, DOI: 10.1002/chem.200400091.
(2) Bachrach, S. M. "DFT Study of [2.2]-, [3.3]-, and [4.4]Paracyclophanes: Strain Energy, Conformations, and Rotational Barriers," J. Phys. Chem. A 2011, 115, 2396-2401, DOI: 10.1021/jp111523u.
(3) Dodziuk, H.; Szymański, S.; Jaźwiński, J.; Ostrowski, M.; Demissie, T. B.; Ruud, K.; Kuś, P.; Hopf, H.; Lin, S.-T. "Structure and NMR Spectra of Some [2.2]Paracyclophanes. The Dilemma of [2.2]Paracyclophane Symmetry," J. Phys. Chem. A 2011, 115, 10638-10649, DOI: 10.1021/jp205693a.
(4) Wolf, H.; Leusser, D.; R. V. Jørgensen, M.; Herbst-Irmer, R.; Chen, Y.-S.; Scheidt, E.-W.; Scherer, W.; Iversen, B. B.; Stalke, D. "Phase Transition of [2,2]-Paracyclophane – An End to an Apparently Endless Story," Chem. Eur. J. 2014, 20, 7048–7053, DOI: 10.1002/chem.201304972.
InChIs
1: InChI=1S/C16H16/c1-2-14-4-3-13(1)9-10-15-5-7-16(8-6-15)12-11-14/h1-8H,9-12H2
InChIKey=OOLUVSIJOMLOCB-UHFFFAOYSA-N