Compounds like antipyrine 1 might be expected to have two pyramidal nitrogens with their substituents on opposite sides of the ring. Interestingly, a new polymorph of propyphenazone 2 has both N-methyl and N-phenyl groups on the same side of the ring. Just how unusual is this?
|
|
|
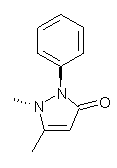
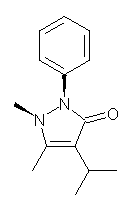
Roumanos and Kertesz1 have sampled the crystallographic database and found 334 structures with the antipyrine backbone. The vast majority of them have the nitrogen substituents on opposite sides, and a few structures have these groups essential co-planar with the ring. The new propyphenazone structure does seem to be unusual. However, they also performed a BLYP/DNP scan of the potential energy surface of 2. When this surface is overlayed on the distribution of the x-ray structures, one sees that most structures are within 3 kcal mol-1 of the energy minimum (with the nitrogen groups on opposite sides). However, the structure with both groups on the same side is about 4 kcal mol-1 higher in energy than the minimum energy structure, and the nearly planar structures are higher in energy still. Thus, the authors conclude that while this new structure is unusual, it is not an outlier.
References
(1) Roumanos, M.; Kertesz, M., "Conformations of Antipyrines," J. Phys. Chem.A, 2011, ASAP, DOI: 10.1021/jp201510w
InChIs
1: InChI=1/C11H12N2O/c1-9-8-11(14)13(12(9)2)10-6-4-3-5-7-10/h3-8H,1-2H3
InChIKey=VEQOALNAAJBPNY-UHFFFAOYAS
2: InChI=1/C14H18N2O/c1-10(2)13-11(3)15(4)16(14(13)17)12-8-6-5-7-9-12/h5-10H,1-4H3
InChIKey=PXWLVJLKJGVOKE-UHFFFAOYAH

Henry Rzepa responded on 10 Jun 2011 at 1:04 am #
A technique which perhaps computational chemists do not use enough comes from the Cambridge crystal database, via a program called Mogul. One can drop (computationally derived) coordinates into it. Mogul then compares all bond lengths, angles and torsions with the ENTIRE crystal database (~600,000 structures), with histogram plots for any one that you wish to view. One can, within seconds, see exactly how much of an outlier any calculation is.
However, a significant limitation of Mogul is that it does not include hydrogen coordinates in its comparisons (on the grounds that the quality of the data for this element in the database is highly variable). Were it not for the fact that the CCDC database is commercial, I would even advocate that the workflow for computational modelling include an automatic pass through the equivalent of Mogul. Peter Murray-Rust has demonstrated such workflows (for example CrystalEye), although of course no open collection of crystal structures has quite the mass of the CCDC (yet!). Perhaps, as the movement for open data grows, we can start routine planning for such workflows. Part of the Semantic Web vision is to populate the web with such data, thus enabling these sorts of validations to be done routinely (i.e.by machines, who do not get bored, unlike humans). It should (to quote a famous computer entrepreneur) “just work”.