Dynamic effects rear up yet again in a seemingly simple reaction. Singleton has examined the Diels-Alder cycloaddition of acrolein with methyl vinyl ketone to give two cross products 1 and 2.1 Upon heating the product mixture, 1 is essentially the only observed species. The retro-Diels-Alder is much slower than the conversion of 2 into 1. Using a variety of rate data, the best estimate for the relative formation of 1:2 is 2.5.

The eight possible transition states for this reaction were computed with a variety of methodologies, all providing very similar results. The lowest energy TS is TS3. A TS of type TS4 could not be found; all attempts to optimize it collapsed to TS3.

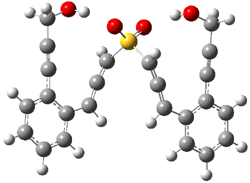
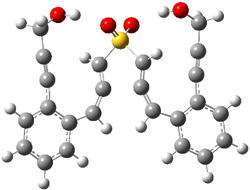
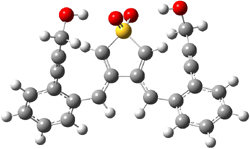
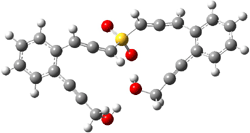
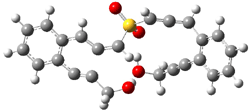
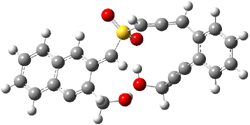
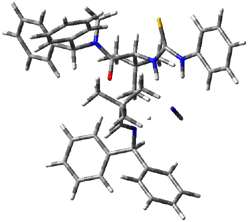
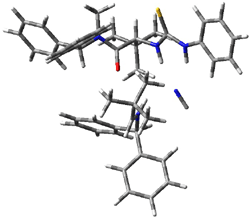
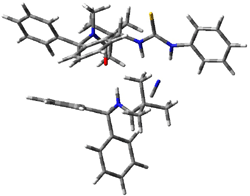
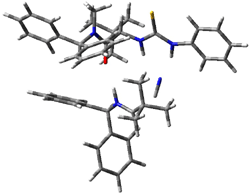
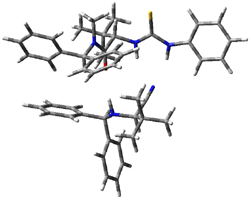
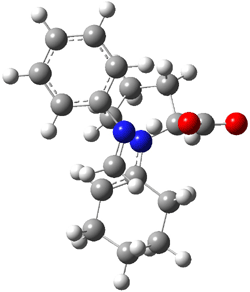
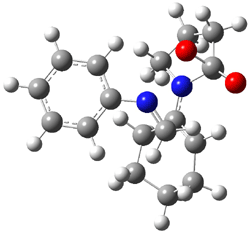
IRC computations indicate the TS3 leads to 1. The lowest energy TS that leads to 2 is TS6, but a second TS (TS5) lower in energy than TS6 also leads to 1. The other TS are still higher in energy. A Cope-type TS that interconverts 1 and 2 (TS7) was also located. The geometries of these TSs are shown in Figure 1.
 TS3 |
 TS5 |
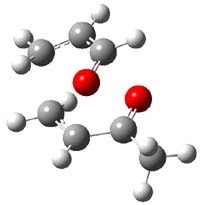
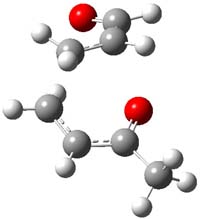 TS6 |
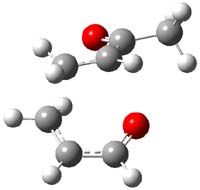
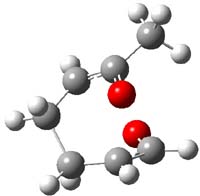 TS7 |
Figure 1. MP2/6-311+G** optimized geometries and relative energies (kcal mol-1) of TS3-TS7.1
Ordinary transition state theory cannot explain the experimental results – the energy difference between the lowest barrier to 1 (TS3) and to 2 (TS6) suggests a rate preference of over 700:1 for 1:2. But the shape of the potential energy surface is reminiscent of others that have been discussed in both my book (Chapter 7) and this blog (see my posts on dynamics) – a surface where trajectories cross a single TS but then bifurcate into two product wells.
To address the chemical selectivity on a surface like this, one must resort to molecular dynamics and examine trajectories. In their MD study of the 296 trajectories that begin at TS3 with motion towards product, 89 end at 1 and 33 end at 2, an amazingly good reproduction of experimental results! Interestingly, 174 trajectories recross the transition state and head back towards reactants. These recrossing trajectories result from “bouncing off” the potential energy wall of the forming C4-C5 bond.
In previous work, selectivity in on these types of surfaces was argued in terms of which well the TS was closer to. But analysis of the trajectories in this case revealed that a strong correlation exists between the initial direction and velocity in the 98 cm-1 vibration – the vibration that corresponds to the closing of the second σ bond, the one between C6-O1 (forming 1), in the negative direction, and closing the C3-O8 bond (forming 2) in the positive direction. Singleton argues that this is a type of dynamic matching, and it might be more prevalent that previously recognized.
References
(1) Wang, Z.; Hirschi, J. S.; Singleton, D. A., "Recrossing and Dynamic Matching Effects on Selectivity in a Diels-Alder Reaction," Angew. Chem. Int. Ed., 2009, 48, 9156-9159, DOI: 10.1002/anie.200903293
InChIs
1: InChI=1/C7H10O2/c1-6(8)7-4-2-3-5-9-7/h3,5,7H,2,4H2,1H3
InChIKey=AOFHZPHBPUYLAG-UHFFFAOYAJ
2: InChI=1/C7H10O2/c1-6-3-2-4-7(5-8)9-6/h3,5,7H,2,4H2,1H3
InChIKey=PLZQHPPETMMEED-UHFFFAOYAD