Houk has performed a very nice examination of the performance of some density functionals.1 He takes a quite different approach than what was proposed by Grimme – the “mindless” benchmarking2 using random molecules (see this post). Rather, Houk examined a series of simple aldol, Mannich and α-aminoxylation reactions, comparing their reaction energies predicted with DFT against that predicted with CBQ-QB3. The idea here is to benchmark DFT performance for simple reactions of specific interest to organic chemists. These reactions are of notable current interest due their involvement in organocatalytic enantioselective chemistry (see my posts on the aldol, Mannich, and Hajos-Parrish-Eder-Sauer-Wiechert reaction). Examples of the reactions studied (along with their enthalpies at CBS-QB3) are Reaction 1-3.
|
|
Reaction 1 |
|
|
Reaction 2 |
|
|
Reaction 3 |
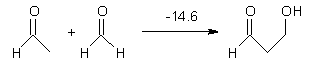
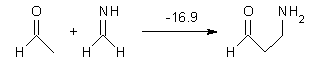
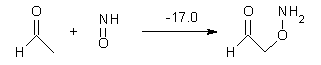
For the four simple aldol reactions and four simple Mannich reactions, PBE1PBE,
mPW1PW91 and MO6-2X all provided reaction enthalpies with errors of about 2 kcal mol-1. The much maligned B3LYP functional, along with B3PW91 and B1B95 gave energies with significant larger errors. For the three α-aminoxylation reactions, the errors were better with B3PW91 and B1B95 than with PBE1PBE or MO6-2X. Once again, it appears that one is faced with finding the right functional for the reaction under consideration!
Of particular interest is the decomposition of these reactions into related isogyric, isodesmic
and homdesmic reactions. So for example Reaction 1 can be decomposed into Reactions 4-7 as shown in Scheme 1. (The careful reader might note that these decomposition reactions are isodesmic and homodesmotic and hyperhomodesmotic reactions.) The errors for Reactions 4-7 are typically greater than 4 kcal mol-1 using B3LYP or B3PW91, and even with MO6-2X the errors are about 2 kcal mol-1.
Scheme 1.

Houk also points out that Reactions 4, 8 and 9 (Scheme 2) focus on having similar bond changes as in Reactions 1-3. And it’s here that the results are most disappointing. The errors produced by all of the functionals for Reactions 4,8 and 9 are typically greater than 2 kcal mol-1, and even MO2-6x can be in error by as much as 5 kcal mol-1. It appears that the reasonable performance of the density functionals for the “real world” aldol and Mannich reactions relies on fortuitous cancellation of errors in the underlying reactions. Houk calls for the development of new functionals designed to deal with fundamental simple bond changing reactions, like the ones in Scheme 2.
Scheme 2

References
(1) Wheeler, S. E.; Moran, A.; Pieniazek, S. N.; Houk, K. N., "Accurate Reaction Enthalpies and Sources of Error in DFT Thermochemistry for Aldol, Mannich, and α-Aminoxylation Reactions," J. Phys. Chem. A 2009, 113, 10376-10384, DOI: 10.1021/jp9058565
(2) Korth, M.; Grimme, S., ""Mindless" DFT Benchmarking," J. Chem. Theory Comput. 2009, 5, 993–1003, DOI: 10.1021/ct800511q

Henry Rzepa responded on 04 Mar 2010 at 1:18 am #
The proliferation of functionals reminds me of that other semiempirical approach introduced by Michael Dewar. Nowadays we have a variety of parameter sets optimised for a purpose. Thus we have parameters for the biochemically important elements (sponsored by eg the NIH), parameters for transition state surfaces, parameters for accurately modelling hydrogen bonding and most recently, parameters for including dispersion corrections.
I suppose there are two kinds of purposes. Those where one interpolates chemistry and those where one extrapolates chemistry. Perhaps this is matched by two kinds of scientist, interpolative and extrapolative (I am of course being provocative!). There is, I would contend, even a third kind of chemist, and they come from the planet Zog. We need all three types!