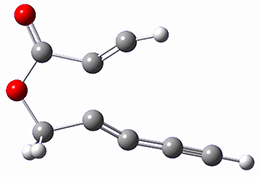
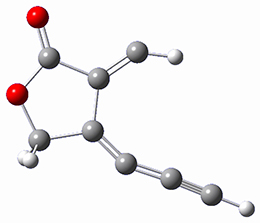
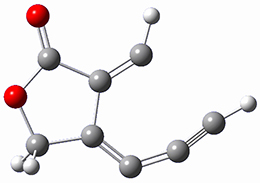
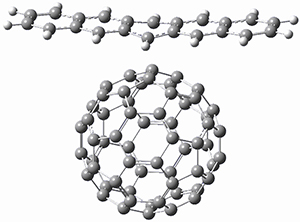
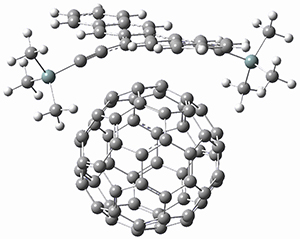
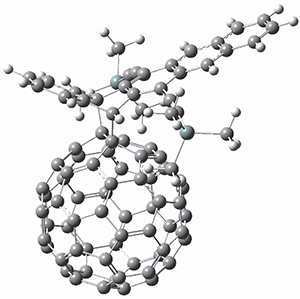
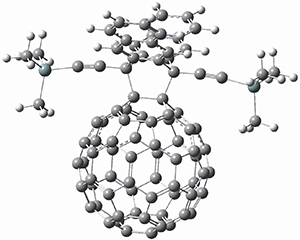
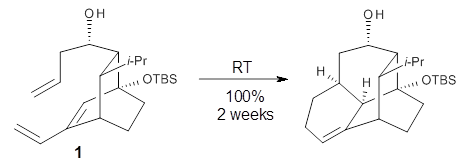
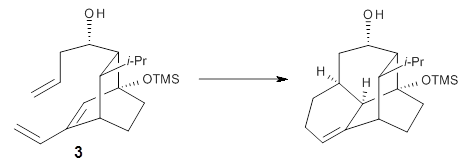
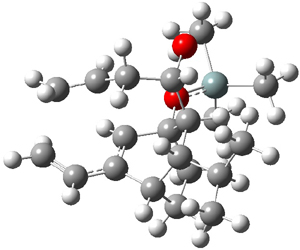
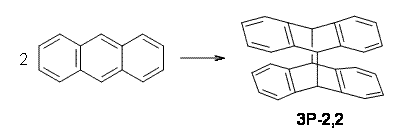
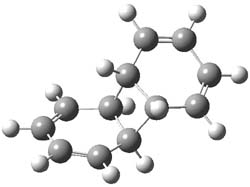
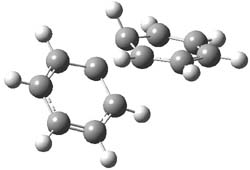
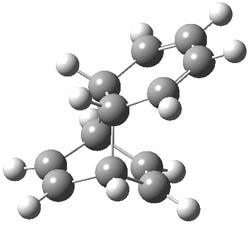
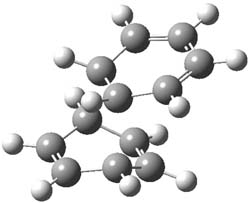
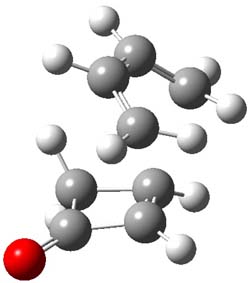
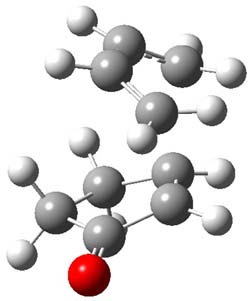
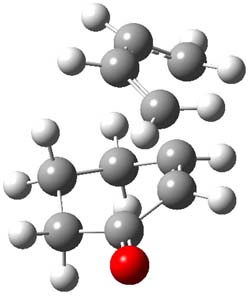
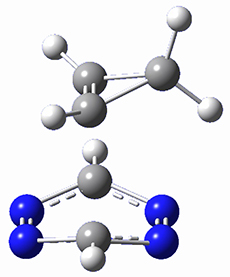
Houk and Doubleday report yet another example of dynamic effects in reactions that appear to be simple, ordinary organic reactions.1 Here they look at the Diels-Alder reaction of tetrazine 1 with cyclopropene 2. The reaction proceeds by first crossing the Diels-Alder transition state 3 to form the intermediate 4. This intermediate can then lose the anti or syn N2, through 5a or 5s, to form the product 6. The structures and relative energies, computed at M06-2X/6-31G(d), of these species are shown in Figure 1.
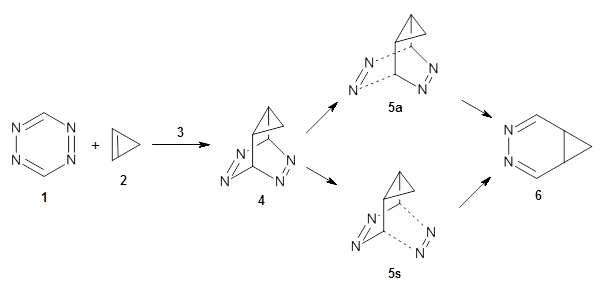
3 |
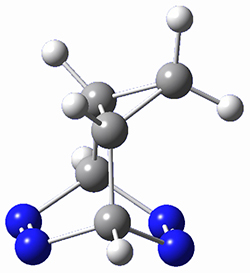
4 |
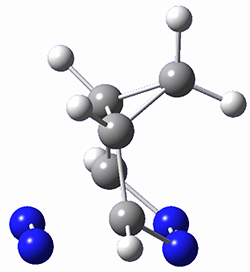
5a |
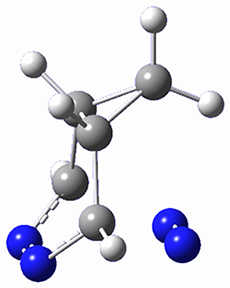
5s |
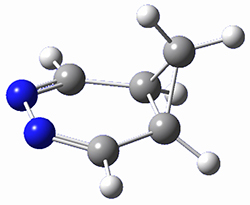
6 |
|
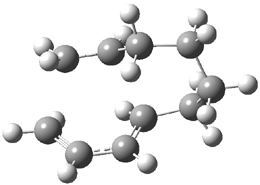
Figure 1. M06-2X/6-31G(d) optimized geometries and energies (relative to 1 + 2) of the critical points along the reaction of tetrazine with cyclopropene.
The large difference in the activation barriers between crossing 5a and 5s (nearly 9 kcal mol-1) suggests, by transition state theory, a preference of more than a million for loss of the anti N2 over the syn N2. However, quasiclassical trajectory studies, using B3LYP/6-31G(d), finds a different situation. The anti pathway is preferred, but only by a 4:1 ratio! This dynamic effect arises from a coupling of the v3 mode which involves a rocking of the cyclopropane ring that brings a proton near the syn N2 functionality, promoting its ejection. In addition, the trajectory studies find short residence times within the intermediate neighborhood for the trajectories that lead to the anti product and longer residence times for the trajectories that lead to the syn product. All together, a very nice example of dynamic effects playing a significant role in a seemingly straightforward organic reaction.
References
(1) Törk, L.; Jiménez-Osés, G.; Doubleday, C.; Liu, F.; Houk, K. N. "Molecular Dynamics of the Diels–Alder Reactions of Tetrazines with Alkenes and N2 Extrusions from Adducts," J. Am. Chem. Soc. 2015, 137, 4749-4758, DOI: 10.1021/jacs.5b00014.
InChIs
1: InChI=1S/C2H2N4/c1-3-5-2-6-4-1/h1-2H
InChIKey=HTJMXYRLEDBSLT-UHFFFAOYSA-N
2: InChI=1S/C3H4/c1-2-3-1/h1-2H,3H2
InChIKey=OOXWYYGXTJLWHA-UHFFFAOYSA-N
4: InChI=1S/C5H6N4/c1-2-3(1)5-8-6-4(2)7-9-5/h2-5H,1H2
InChIKey=JGSMBFYJCNPYDM-UHFFFAOYSA-N
6: InChI=1S/C5H6N2/c1-4-2-6-7-3-5(1)4/h2-5H,1H2
InChIKey=RYJFHKGQZKUXEH-UHFFFAOYSA-N