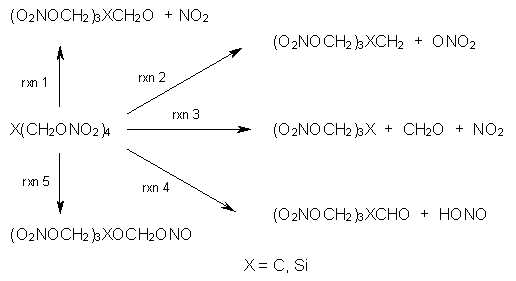
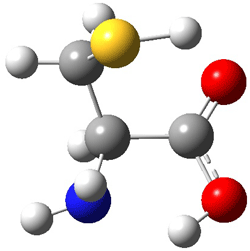
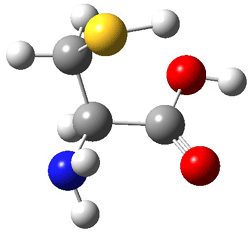
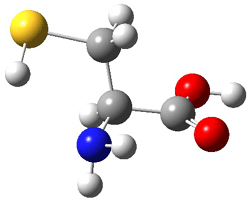
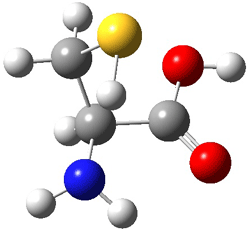
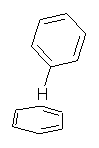
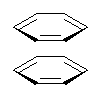
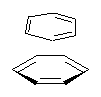
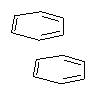
Here’s another fine paper from the Alonso group employing laser ablation molecular beam Fourier transform microwave spectroscopy coupled with computation to discern molecular structure. In this work they examine the low-energy tautomers of guanine.1 The four lowest energy guanine tautomers are shown in Figure 1. (Unfortunately, Alonso does not include the optimized coordinates of these structures in the supporting information – we need to more vigorously police this during the review process!) These tautomers are predicted to be very close in energy (MP2/6-311++G(d,p), and so one might expect to see multiple signals in the microwave originating from all four tautomers. In fact, they discern all four, and the agreement between the computed and experimental rotational constants are excellent (Table 1), especially if one applies a scaling factor of 1.004. Once again, this group shows the power of combined experiment and computations!
|
|
|
|
|
|
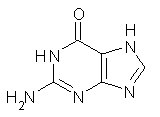
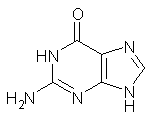
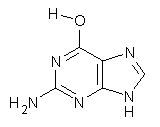
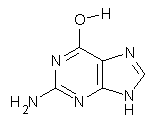
Figure 1. Four lowest energy (kcal mol-1, MP2/6-311++G(d,p)) tautomers of guanine.
Table 1. Experimental and computed rotational constants (MHz) of the four guanine tautomers.
|
|
1 |
2 |
3 |
4 |
||||
|
|
Exp |
Comp |
Exp |
Comp |
Exp |
Comp |
Exp |
Comp |
|
A |
19.22155 |
1909.0 |
19.222780 |
1909.7 |
1916.080 |
1908.6 |
1923.460 |
1915.6 |
|
B |
1121.6840 |
119.2 |
1116.6710 |
1113.5 |
1132.360 |
1128.2 |
1136.040 |
1131.9 |
|
C |
709.0079 |
706.6 |
706.8580 |
704.2 |
712.1950 |
709.5 |
714.7000 |
712.0 |
References
(1) Alonso, J. L.; Peña, I.; López, J. C.; Vaquero, V., "Rotational Spectral Signatures of Four Tautomers of Guanine," Angew. Chem. Int. Ed. 2009, 48, 6141-6143, DOI: 10.1002/anie.200901462
InChIs
Guanine: InChI=1/C5H5N5O/c6-5-9-3-2(4(11)10-5)7-1-8-3/h1H,(H4,6,7,8,9,10,11)/f/h8,10H,6H2
InChIKey=UYTPUPDQBNUYGX-GSQBSFCVCX