Another remarkable piece of science from the Schreiner and Allen groups has appeared demonstrating the critical importance of combining experiment with computations.1 (This one will surely be in the running for computational chemistry paper of the year.) Once again they examine tunneling from a carbene intermediate, but this time with an amazing conclusion that will have impact on chemistry textbooks!
Schreiner and Allen have previously examined a number of hydroxycarbenes (see these posts: A, B, C) and have found tunneling to be the main exit channel from these carbenes. The tunneling passes through barriers that are as large as 30 kcal mol-1, and as expected, the deuterium labeled analogues have tunneling half lives that are exceptionally long, like 4000 years.
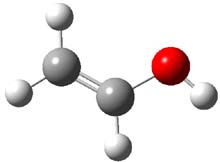
Now they examine methylhydroxycarbene 1,1 which is interesting because there are two possible exit channels, leading to acetaldehyde 2 or vinyl alcohol 3. Previous gas-phase pyrolysis of pyruvic acid suggested the intermediacy of 1, which rearranges to 2 much more rapidly than to 3. However, G1 computations predict the barrier to 3 is smaller than the barrier to 2,2 which should mean that 2 is the kinetic product!

Methylhydroxycarbene 1 was prepared by flash pyrolysis of pyruvic acid with capture of the products in an argon matrix. The carbene 1 was characterized by IR. The predicted frequencies (CCSD(T)/cc-pCVTZ – with corrections for anharmonicity) of 9 of the 11 bands of 1 are within 8 cm-1 of the experimental frequencies. The OH and OD stretches, the ones not in agreement, are likely to be perturbed by the matrix. The predicted (MRCC/aug-cc-pVTZ) and experimental UV spectrum are also in close agreement.
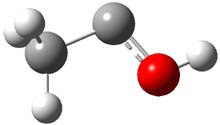
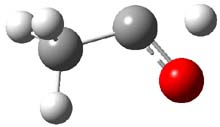
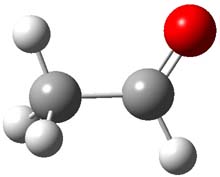
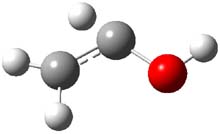
Holding the matrix at 11 K and following the spectra of 1-3 led to the following important kinetic results: the half-life for formation of 2 is 66 min with no 3 observed to form. In addition, the rate for the deuterium labeled carbene to form 2 was too long for measuring, but was 196 minutes in Kr and 251 minutes in Xe. CCSD(T)/cc-pCVCZ computations followed by focal point methods gives the barrier to form acetaldehyde from 1 as 28.0 kcal mol-1 while that to form vinyl alcohol 3 is much lower: 22.6 kcal mol-1. (The structures of these three molecules and the transition states connecting them are shown in Figure 1.) Apparently, the reaction passes through or over the higher barrier in large preference over passing through or over the lower barrier!
Figure 1. CCSD(T)/cc-pCVTZ optimizes structures of 1-3 and the transition states connecting 1 to 2 and 1 to 3.
Precise mapping of the intrinsic reaction path at CCSD(T)/cc-pCVTZ allows for computing the WKB tunneling probabilities. This leads to the prediction of the half-life for the reaction 1 → 2 as 71 minutes, in excellent agreement with experiment. The computed half-life for the deuterium labeled reaction of 400 years and the computed half-life for 1 → 3 of 190 days are both in fine agreement with experiment.
Why does the reaction preferentially tunnel through the higher barrier? Well, the tunneling rate is dependent on the square root of the barrier height and linearly on the barrier width. The width is much smaller for the rearrangement to 2. The hydrogen needs to move a shorter amount in proceeding from 1to 2 than to 3, and in the rearrangement to vinyl alcohol a second hydrogen must migrate downwards to form the planar vinyl group. Basically, width beats out the height.
The important conclusion from this paper is the following: in addition to reactions being under kinetic or thermodynamic control, we must now consider a third options – a reaction under tunneling control!
A nice perspective on this paper and its implications has been written by Carpenter, who points out how this adds to our general notion of significant limitations to transition state theory.3
References
(1) Schreiner, P. R.; Reisenauer, H. P.; Ley, D.; Gerbig, D.; Wu, C.-H.; Allen, W. D., "Methylhydroxycarbene: Tunneling Control of a Chemical Reaction," Science, 2011, 332, 1300-1303, DOI: 10.1126/science.1203761.
(2) Smith, B. J.; Nguyen Minh, T.; Bouma, W. J.; Radom, L., "Unimolecular rearrangements connecting hydroxyethylidene (CH3-C-OH), acetaldehyde (CH3-CH:O), and vinyl alcohol (CH2:CH-OH)," J. Am. Chem. Soc., 1991, 113, 6452-6458, DOI: 10.1021/ja00017a015
(3) Carpenter, B. K., “Taking the High Road and Getting There Before You,” Science, 2011, 332, 1269-1270, DOI: 10.1126/science.1206693.
InChIs
1: InChI=1/C2H4O/c1-2-3/h3H,1H3
InChIKey=JVKQHDUTAFISFX-UHFFFAOYAN
2: InChI=1/C2H4O/c1-2-3/h2H,1H3
InChIKey=IKHGUXGNUITLKF-UHFFFAOYAB
3: InChI=1/C2H4O/c1-2-3/h2-3H,1H2
InChIKey=IMROMDMJAWUWLK-UHFFFAOYAT